
缺氧、缺氧/复氧对心肌细胞内ROS、MAPKs 活性表达的影响
2021-05-15 02:17:56凌学斌綦苗苗李继科郭峻莉李天发
海南医学院学报 2021年7期
凌学斌,王 军,綦苗苗,李继科,郭峻莉,李天发
(海南医学院第一附属医院心血管内科,心血管病研究所,海南海口570102)
细胞凋亡是引起心力衰竭的重要病理机制,常见诱因包括心肌缺血再灌注(ischemia/reper‐fusion,I/R)损伤、慢性压力负荷及充血性心力衰竭[1]。I/R 损伤常见于溶栓、冠状动脉介入或搭桥、心脏移植术等治疗中,是临床上治疗缺血性心脏病的常用方法,但可诱发心肌顿抑、再灌注恶性心律失常、心肌细胞凋亡及心脏收缩和舒 张 功 能 不 全[2]。 研 究 发 现 活 性 氧(reactive oxygen species,ROS)介导的氧化应激可进一步活 化MAPKs 通 路[3],是 心 肌I/R 损 伤 的 重 要 病理 过 程。ROS 包 括O2•−、·OH、ONOO-、H2O2等,目前被认为是细胞内第二信使,影响着细胞增殖、分 化、凋 亡 及 坏 死 等 过 程[4]。MAPKs 为 丝氨酸-苏氨酸蛋白激酶,主要包括细胞外信号调节蛋白激酶(ERK)、氨基末端激酶(JNK)及p38,通过磷酸化下游产物调节细胞的生长、分化、对环境的应激适应、炎症反应。目前有关心肌细胞I/R 损伤的研究常使用具有成熟心肌细胞特性的H9c2 心肌细胞株,通过建立缺氧/复氧(hypoxia/reoxygenation,H/R)模型来完成。H9c2细 胞 经H/R 处 理 后 可 激 活ROS/ERK[3]、ROS/JNK[3,5]及ROS/p38[6]通 路,进 而 诱 导 心 肌 细 胞凋亡。目前尚少有研究关注ROS 对ERK、JNK及p38 活性水平的表达是否存在差异以及缺氧、H/R 对上述通路是否存在影响。本研究通过CoCl2诱导H9c2 缺氧,使用完全培养基模拟I/R损伤病理过程,比较常氧、缺氧与H/R 条件下ROS 水 平、MAPKs 及Caspase-3 活 性 变 化,探 讨ROS/MAPKs 通路对H9c2 心肌细胞氧化应激损伤的作用。
1 材料与方法
1.1 材料与试剂
高糖DMEM 培养基购自hyclone 公司,胎牛血清购自BI 公司,青链霉素购自索莱宝公司,CoCl2·6H2O、兔抗磷酸化JNK(p-JNK)、兔抗p-ERK1/2 购自生工生物工程有限公司,兔抗p-p38、HRP 标记山羊抗兔二抗购自Santa Cruze 公司,细胞裂解液、兔抗α-Tubulin、兔抗Caspase-3、DHE荧光探针购自碧云天、DCFH-DA 荧光探针购自Sigma Aldrich 公司。H9c2 由中国科学院上海生命科学研究院细胞资源中心提供。
1.2 细胞培养及H/R 模型建立
H9c2 心肌细胞株来源于大鼠胚胎期心脏组织,在37 ℃、5% CO2条 件 下 培 养 于 含10% 胎 牛血清的高糖DMEM 培养基中。对照组为完全培养基培养16 h,缺氧组给予终浓度600 μ mol/L CoCl2的DMEM 培养液进行化学模拟缺氧16 h,H/R 组细胞在模拟缺氧后,换不含CoCl2DMEM培养液继续培养4 h。
1.3 细胞存活率检测
H9c2 心肌细胞接种于96 孔培养板中,当细胞生长到培养孔的80% 面积时,根据实验要求进行处理:(1)设置CoCl2浓度梯度(0、150、300、450、600、900、1 200、2 400 μ mol/L)培 养24 h;(2)设置时间梯度(空白、4、12、16、24、48 h)600 μ mol/L CoCl2培养,每组设5 个复孔。上述过程完成后,每孔加10% MTT 200 μ L,培养箱中培养4 h,加DMSO 150 μ L,震荡10 min,最后酶标仪(λ=490 nm)下检测吸光度(OD 值)。取6 孔OD 值的平均数,按公式计算细胞存活率,细胞存活率(%)= OD 处理组/OD 对照组×100%,重复3 次。
1.4 细胞内ROS 水平的测定
将赖氨酸包被的盖玻片置于6 孔培养板内,H9c2 心肌细胞被均匀的接种在盖玻片上。当细胞生长到培养孔约80% 面积时,根据实验需要给予相应的处理:(1)缺氧组600 μ mol/L CoCl2分别处理8、16 h;(2)H/R 组:600 μ mol/L CoCl2分别处理8、16 h 后,换用不含CoCl2DMEM 培养4 h。每组包括3 个复孔,处理完后,用PBS 漂洗2 次,分 别 对H9c2 心 肌 细 用10 μ mol/L 绿 色 荧 光探针DCFH-DA 检测缺氧8 h/复氧4 h、5 μ mol/L 红色荧光探针DHE 检测缺氧16 h/复氧4 h,37 ℃孵育30 min。在荧光显微镜下随机选取5个不重复区摄片,用Image J1.42q 软件进行平均荧光强度(mean flourscence indensity,MFI)分析。
1.5 Western blot 法检测p-p38、p-ERK1/2、p-JNK、Caspase-3 表达
H9c2 心肌细胞接种于35 mm 培养皿内,培养至80% 满时,分别按上述处理条件处理对照组、缺氧组及H/R 组细胞,处理各组细胞后用预冷的PBS 洗2 次,加入细胞裂解液,裂解数分钟后,12 000 r/min 离心10 min,取上清,采用考马斯亮蓝G-250 测定蛋白含量。总蛋白经十二烷基硫酸钠聚丙烯酰胺凝胶电泳分离后,转移到PVDF 膜上。用2% 胎牛血清蛋白封闭2 h,TBS洗3 次,随后加入一抗p-JNK(1∶500)、p-ERK1/2(1∶500)、p-p38(1∶1 000)、Caspase-3(1∶1 000)、α-Tubulin(1∶1 000)4 ℃过 夜,TBST 洗3 次 后 用HRP 标记山羊抗兔(1∶5 000)37 ℃孵育1 h,TBST洗PVDF 膜3 次,用发光试剂ECL 显色,暗室曝光到X 光片上,凝胶成像系统扫描分析结果。
1.6 统计学处理
全部实验数据经SPSS 19 软件进行统计学分析,所有结果以(±s)表示,组间比较采用单因素方差分析(one-way ANOVA)检验,用LSD进行均数之间的比较,P<0.05 为差异有统计学意义。
2 结果
2.1 CoCl2呈浓度依赖性及时间依赖性抑制H9c2 心肌细胞成活率
图1A 及表1 显示,与对照组相比,不同浓度的CoCl2处 理H9c2 心 肌 细 胞16 h 后,150 μ mol/L CoCl2对H9c2 心 肌 细 胞 活 力 无 影 响,从300 μ mol/L 开始呈剂量依赖性抑制心肌细胞活力(P<0.01)。与 前 一 梯 度 浓 度 相 比,在600 μ mol/L 处下降最为明显(P<0.001),随后1200 μ mol/L 与900 μ mol/L、2 400 μ mol/L 与1 200 μ mol/L 细胞活力差别无统计学意义,可能与细胞处于生长对数期抵制CoCl2毒性作用有关。图1B 及表2 显示不同时间段(0~48 h)下,与对照组相比,300、600 及2400 μ mol/L 的CoCl2均 呈 时 间 依 赖 性 抑 制H9c2 心肌 细 胞 活 力(P<0.01),其 中300、600 μ mol/L 在12~24 h 内细胞活力差别无统计学意义,24 h 后显著下降(P<0.001),而2 400 μ mol/L 的CoCl2除12~16 h 外细胞活力差别均存在显著差异(P<0.001)。综上,为有效抑制细胞活力并减少细胞过 多 的 凋 亡,选 择600 μ mol/L 的CoCl2培 养16 h。
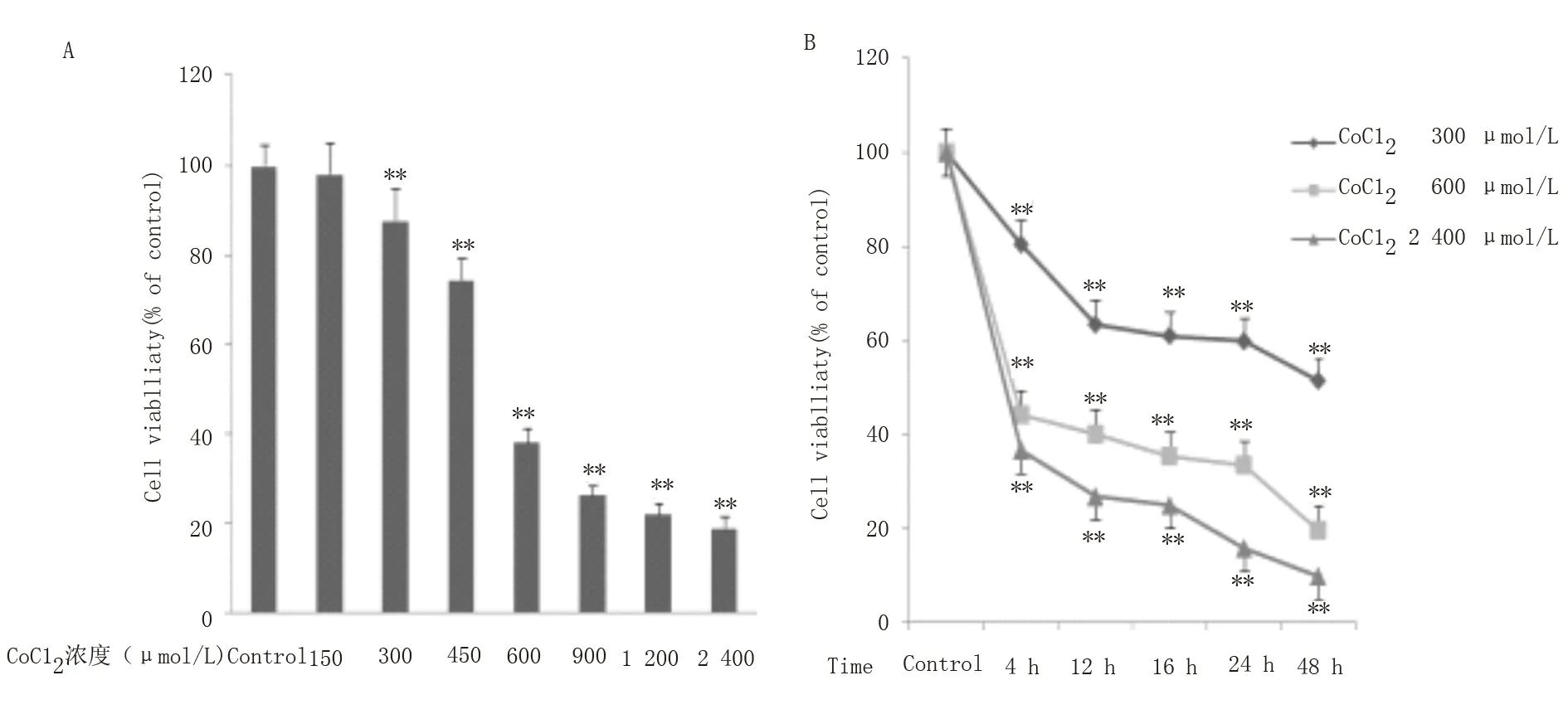
图1 CoCl2对H9c2 心肌细胞存活率的影响Fig 1 Effect of CoCl2 on H9c2 viability
表1 不同浓度CoCl2 处理16 h 对H9c2 心肌细胞存活率的影响(±s)Tab 1 Effect of different concentrations of CoCl2 for 16 h on survival rate of H9c2(±s)

表1 不同浓度CoCl2 处理16 h 对H9c2 心肌细胞存活率的影响(±s)Tab 1 Effect of different concentrations of CoCl2 for 16 h on survival rate of H9c2(±s)
OD 值OD CoCl2(μmol/L)0 100.0±4.5 t P--150 98.1±6.8 1.47 0.265 300 87.8±6.9 12.92 0.005 450 74.5±4.8 90.68<0.001 600 38.2±3.1 771.76<0.001 900 26.3±2.2 1 309.00<0.001 1 200 22.2±2.5 1 380.00<0.001 2 400 19.0± 2.5 1 491.00<0.001
表2 300、600 及2 400 μmol/L CoCl2时在不同时间段对H9c2 心肌细胞存活率的影响(%,±s)Tab 2 Effects of CoCl2 at 300,600 and 2 400 μmol/L on survival rate of H9c2 cardiomyocytes at different times(%,±s)

表2 300、600 及2 400 μmol/L CoCl2时在不同时间段对H9c2 心肌细胞存活率的影响(%,±s)Tab 2 Effects of CoCl2 at 300,600 and 2 400 μmol/L on survival rate of H9c2 cardiomyocytes at different times(%,±s)
组别300 μmol/L CoCl2 组600 μmol/L CoCl2 组2 400 μmol/L CoCl2 组0 h 99±2.2 100±4.5 100±3.3 4 h 80.5±6.7 44.2±8.2 36.6±4.8 12 h 63.4±0.3 40.1±2.5 26.7±3.3 16 h 60.9±0.9 35.3±1.7 24.9±3.3 24 h 59.8±2.7 33.5±9.6 15.8±1.1 48 h 51.3±0.8 19.6±0.9 9.8±0.8 F P 20.17 632.00 286.00<0.001<0.001<0.001
2.2 ROS 水平与H9c2 心肌细胞 缺氧、H/R 关系
图2 显 示 了ROS 荧 光 探 针 检 测600 μ mol/L的CoCl2不同时间处理下H9c2 心肌细胞缺氧、缺氧/复ROS 水平变化。与对照组相比,缺氧4 h后采用DCFH-DA 活性氧探针MFI 为其1.9 倍(P<0.05),缺氧4 h/复氧4 h 后MFI 为 其4.2 倍(P<0.001);缺氧16 h 后采用DHE 超氧化物阴离子荧光探针MFI 为其1.6 倍(P<0.0 1),缺氧1 6 h/复氧4 h MFI 为其2.5 倍(P<0.001);缺氧4 h 或16 h 后复氧均可使ROS 水平产生进一步增多(P<0.01),见表3。DCFH-DA 和DHE 探针法检测结果表明CoCl2诱导的化学性缺氧剂可导致H9c2 心肌细胞内ROS 水平升高,去除CoCl2后模拟缺氧复氧环境仍可促进ROS 表达。
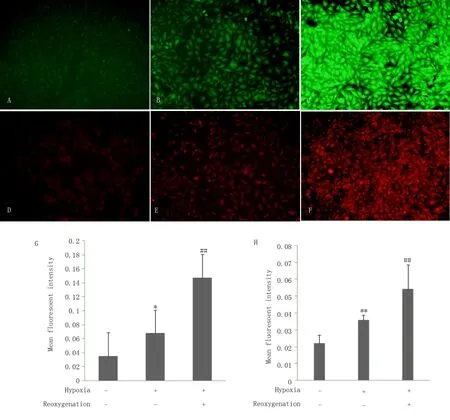
图2 缺氧、H/R 对H9c2 细胞内ROS 水平的影响Fig 2 Effect of hypoxia and hypoxia/reoxygenation on ROS in H9c2
2.3 缺氧、H/R 下H9c2 心肌细胞内p-JNK、p-ERK1/2 和p-p38 蛋白表达影响
结 果 显 示H9c2 心 肌 细 胞 在600 μ mol/L 的CoCl2诱 导 缺 氧16 h,复 氧4 h 时p-MAPKs 活 性变化。蛋白印迹检测显示,与对照组比较,p-JNK和p-p38 表达升高(P<0.05),而p-ERK 变化无统计 学 意 义,提 示600 μ mol/L 的CoCl2诱 导 缺 氧16 h 后 可 引 起p-JNK 和p-p38 明 显 升 高,而p-ERK 无明显变化,当细胞发生H/R 后,与缺氧组比较,三者均明显升高(P<0.01),提示细胞H/R 加重细胞氧化应激损伤,进一步活化p-MAPKs,见图3,表4。
表3 缺氧、H/R 对H9c2 细胞内ROS 水平的影响(±s)Tab 3 Effect of hypoxia and hypoxia/reoxygenation on ROS in H9c2(±s)

表3 缺氧、H/R 对H9c2 细胞内ROS 水平的影响(±s)Tab 3 Effect of hypoxia and hypoxia/reoxygenation on ROS in H9c2(±s)
注:与对照组比较,*P<0.05;与缺氧组比较,##P<0.01。
组别DHE(16 h)对照组缺氧组H/R 组DCFH-DA(4 h)0.035±0.006 0.068±0.015 0.147±0.034 P -P -0.001*<0.001##0.038*<0.001##0.022±0.005 0.036±0.003 0.054±0.006
2.4 H9c2 心肌细胞缺氧、H/R 时对Cleaved Caspase-3 活性的影响
图4 为H9c2 细 胞 在600 μ mol/L 的CoCl2诱 导缺氧16 h,复氧4 h 下细胞凋亡情况。图中无活性Caspase-3 在缺氧、H/R 逐渐减少,转化为有活性的Cleaved Caspase-3 并逐渐增多。从结果中可看出细胞缺氧下可激活Caspase-3 介导的凋亡通路,H/R 后可使该通路进一步活化,见表5。
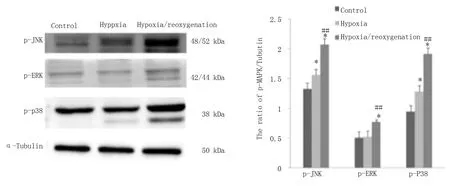
图3 缺氧、H/R 下H9c2 细胞内p-JNK、p-ERK 及p-p38 表达变化Fig 3 Effect of hypoxia and hypoxia/reoxygenation on p-JNK,p-ERK and p-p38 in H9c2
表4 缺氧、H/R 下H9c2 细胞内p-JNK、p-ERK 及p-p38 表达变化(±s)Tab 4 Effects of hypoxia and hypoxia/reoxygenation on p-JNK,p-ERK and p-p38 in H9c2(±s)

表4 缺氧、H/R 下H9c2 细胞内p-JNK、p-ERK 及p-p38 表达变化(±s)Tab 4 Effects of hypoxia and hypoxia/reoxygenation on p-JNK,p-ERK and p-p38 in H9c2(±s)
注:与对照组比较,*P<0.05;与缺氧组比较,##P<0.01。
组别对照组缺氧组H/R 组P-JNK 1.323±0.041 1.559±0.075 2.068±0.070 P-P-P-0.014*<0.001##0.026*<0.001##P-ERK 0.507±0.069 0.524±0.067 0.771±0.066 0.793*0.005##P-P38 0.946±0.024 1.282±0.167 1.914±0.076

图4 缺氧、H/R 下对H9c2 细胞内Caspase-3 活性裂解片段表达的影响Fig 4 Effects of hypoxia and hypoxia/reoxygenation on the expression of cleaved Caspase-3 in H9c2
表5 缺氧、H/R 下对H9c2 细胞内Caspase-3 活性裂解片段表达的影响(±s)Tab 5 Effects of hypoxia and hypoxia/reoxygenation on the expression of cleaved Caspase-3 in H9c2(±s)
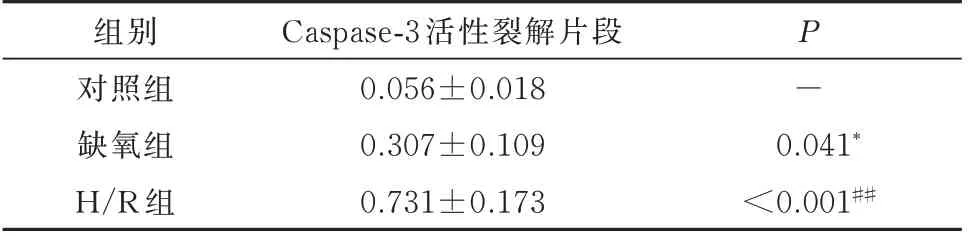
表5 缺氧、H/R 下对H9c2 细胞内Caspase-3 活性裂解片段表达的影响(±s)Tab 5 Effects of hypoxia and hypoxia/reoxygenation on the expression of cleaved Caspase-3 in H9c2(±s)
组别对照组缺氧组H/R 组Caspase-3 活性裂解片段0.056±0.018 0.307±0.109 0.731±0.173 P-0.041*<0.001##
3 讨论
H9c2 细胞株来源于大鼠胚胎心脏组织,具有成熟心肌细胞特性,广泛用于细胞形态、电生理及毒理研究[5]。细胞体外H/R 模型可通过物理性及化学性诱导。物理性缺氧常在充有氮气缺氧装置及常氧装置中完成[6],因需要严格检测氧浓度,技术性要求高所以其运用受到限制。化学性缺氧则操作方便,CoCl2为常用的是化学性低氧模拟剂。钴可替代亚铁螯合于血红蛋白中,使细胞摄氧障碍而引起氧化应激损伤,去除CoCl2溶液换用新鲜培养基可恢复血红蛋白摄氧,引起细胞H/R 损伤[7],进而模拟细胞I/R 损伤。本实验再次证实,CoCl2呈浓度依赖性及时间依赖 性 抑 制H9c2 心 肌 细 胞 成 活 率,与Gallo 等[8]报道的相一致。本研究显示心肌细胞缺氧后可导致ROS 大量产生,而H/R 后ROS 进一步增多,进而证实心肌I/R 损伤与ROS 诱导的氧化应激损伤密切相关。细胞经I/R 处理后产生的ROS可作为第二信使从细胞膜转运到胞浆,并作为第三信使转进入细胞核中,通过与DNA 结合及改变下游基因表达水平引起细胞I/R 损伤,因而目前常用的抗氧化剂如谷胱甘肽、超氧化物歧化酶、辅酶Ⅱ和血红素加氧酶-1,可减少细胞内ROS 水平达到抗氧化应激损伤作用[9]。
H9c2 在H/R 诱导下通过ROS/JNK/Egr-1 通路引发细胞发生氧化应激损伤[10],而腺苷酸活化蛋白激酶的激活可通过JNK 介导的NF-κ B 途径抑制缺氧/复氧时的炎症反应[11]。H9c2 细胞凋 亡 与ROS 激 活JNK/p38 通 路 有 关,Jantira 等[12]证实使用P38 抑制剂可阻断P38 通路进而对糖尿病缺血性心肌病患者起到积极的心脏保护作用。P38 通路在细胞缺血缺氧时可被活化,但并不是出现在所有缺血缺氧模型中,这与细胞何时被激活以及缺血持续时间密切相关[13]。本研究发现,H9c2 在缺氧及H/R 不同处理后JNK 和p38 磷酸化水平均明显升高,而p-ERK 在缺氧处理后升高不明显,但经H/R 后表达水平明显升高,这 与Mizukanmi 等[14]在 对 大 鼠 心 脏I/R 体 内实验结果相一致,该研究发现缺血不足以激活ERK 上 游 分 子MEK(MAP kinase/extracellular signal-regulatedkinase kinase)-2,只有经I/R 处理后可使得MEK-2 磷酸化进而激活ERK。此外,是否需ROS 大量蓄积后才能激活ERK 通路尚需要进一步证实。总之,MAPKs 通路的活化随着ROS不同诱导条件而存在差异,提示三条通路不是独立存在,而是相互作用、相互影响。
在缺氧或H/R 条件下产生的ROS 可诱导心肌细胞氧化应激损伤,主要表现在线粒体、内质网应激、溶酶体等亚细胞结构损伤,可分别通过JNK/ERK/p38 通路介导下诱导细胞凋亡[15-17],本课题组既往研究对此有详细阐释[18]。本研究进一步证实H9c2 经I/R 损伤后可引发更多的心肌细胞发生凋亡。目前ERK 信号通路在细胞凋亡中的作用存在争论,该通路活化既可导致细胞发生凋亡[19],也可抑制肿瘤细胞凋亡使其存活的 作 用[20]。庞 伟[21]同 样 证 实 在 缺 锌 致 海 马 神经细胞氧化应激损伤中发现ERK 活性表达下降,引发细胞凋亡,而适量补锌可通过活化MEK/ERK 信号通路,减缓海马神经细胞损伤。JNK、ERK 及p38 在细胞凋亡中的作用机制尚需要进一步研究。综上所述,CoCl2诱导缺氧、H/R 损伤模型,可模拟心肌细胞缺血、I/R 损伤病理过程,为后续抗氧化应激损伤机制研究提供理论基础和实验依据。
猜你喜欢
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
建材发展导向(2021年11期)2021-07-28 06:57:22
当代水产(2020年10期)2020-03-17 07:02:48
当代水产(2019年8期)2019-10-12 08:57:26
中成药(2018年5期)2018-06-06 03:11:46
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
物理实验(2015年9期)2015-02-28 17:36:47
天然产物研究与开发(2014年6期)2014-04-27 14:16:05
