
I型先天性厚甲症一例
2021-05-11 12:40刘忠艳王启华周桂芝
中国麻风皮肤病杂志 2021年5期
刘忠艳 王启华 周桂芝
1临沂市皮肤病医院皮肤科,临沂市皮肤病防治所,临沂,276000;2山东第一医科大学附属皮肤病医院(山东省皮肤病医院,山东省皮肤病性病防治研究所),济南,250022
临床资料患儿,女,11岁。因“厚甲、掌跖角化11年”于2019年3月来我院门诊就诊。患儿出生2个月发病。鼻孔、口角处起红斑、渗出、结黄痂。肘膝部、臀部、掌跖等摩擦部位易反复出现疼痛和水疱,之后躯干四肢出现皮肤干燥,散在毛囊性角化性丘疹及斑块,掌跖也逐步出现角化。随年龄增长,全部指(趾)甲逐渐呈黄褐色增厚,近几年甲远端翘起,变为褐色。目前,跖趾部摩擦部位仍可出现水疱,易破溃、流渗液。清洗不及时散发臭味。
患者为独生女,顺产,无外伤。声音无嘶哑,头发、牙齿无异常,视力正常,智力可。家族内无结核、肝炎等传染病史。其父45岁,有类似皮肤角化、甲增厚病史(图1),智力正常;其母健康,无类似疾病。其祖父母、外祖父母均体健,无遗传病史,均非近亲结婚。其祖父母生育3个孩子,1子即其父;2女及其后代均体健。其外祖父母生育1子1女,子女及后代均体健。
体格检查:发育正常,营养中等,系统检查未见异常。皮肤科查体:双口角结黄痂,口唇线状白斑,口腔片状白斑,舌苔厚白(图2)。躯干四肢皮肤干燥,散在毛囊性角化性丘疹及斑块,手部以及掌跖部皮肤点状、片状角化,指(趾)甲甲板明显增厚,部分趾甲间有糜烂(图3、4)。牙齿及头发未见异常,未见表皮囊肿和多发性脂囊瘤、皮肤色素沉着。实验室检查:甲板真菌镜检、培养(-)。皮肤组织病理(腰背部)示:表皮角化过度、角化不全、棘层增厚,真皮乳头血管扩张、充血,周围少许组织细胞浸润(图5)。诊断:I型先天性厚甲症。
讨论先天性厚甲症(pachyonychia congenita, PC)是一种以甲增厚性营养不良为特点的外胚叶发育不良,是常染色体显性遗传病[1]。临床上分为四型:I型又称杰达斯索恩-列文道斯基综合征(Jadassohn-lewandowsky syndrome),最为常见。特征:①出生时或出生后不久所有指(趾)甲变厚、变色,常可见甲板脱落;②掌跖角化,呈小片状,少数可完全角化;③大疱,易发生在胼胝下;④口腔黏膜白斑;⑤声音嘶哑;⑥毛发异常,如多毛、扭曲及其他毛发营养不良的表现;⑦掌跖多汗。II型又称杰克逊-劳勒综合征(Jackson-Lawler syndrome),除I型症状外,尚有胎生牙及多发性囊肿。此型一般无口腔黏膜白斑。III型罕见,厚甲及掌跖角化较轻,有角膜白斑、白内障等。IV型除III型症状外,还有咽喉损害、智力障碍及色素沉着等[2]。其中,I型和II型在临床中最为常见。本例患儿为PC-I,具有自幼发病的特点;手足指趾甲持续变色增厚;掌跖片状角化;躯干四肢等部位毛囊角化性丘疹及斑块;黏膜白斑;未见牙齿及头发异常、未见表皮囊肿、多发性脂囊瘤、皮肤色素沉着等临床特征。
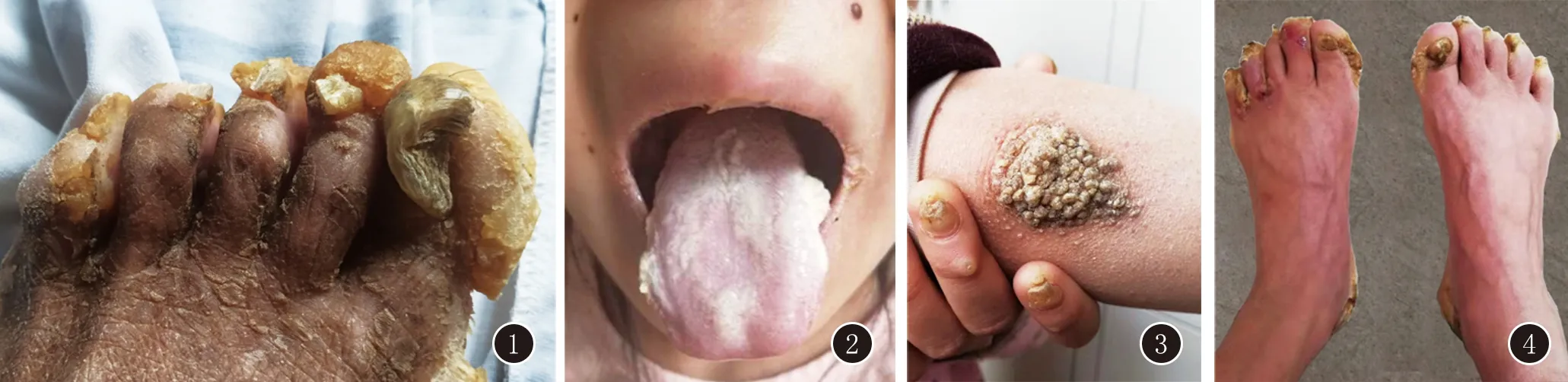
图1 患儿父亲足部皮肤角化过度,趾甲增厚图2 患儿双口角结黄痂,口唇线状白斑,口腔片状白斑,舌苔厚白
图3 患儿皮肤干燥,肘部毛囊性角化性丘疹融合成斑块;指甲甲板增厚图4 患儿趾甲甲板增厚,甲远端翘起,呈褐色;趾侧、趾间渗液、结黄痂

图5 表皮角化过度、角化不全、棘层增厚,真皮乳头血管扩张、充血,周围少许组织细胞浸润(HE,×100)
先天性厚甲症涉及五个角蛋白基因突变,分别为KRT6A、KRT6B、KRT6C、KRT16和KRT17[3]。I型为KRT6a和KRT16突变,II型为KRT6b和KRT17突变[1]。限于我院条件,本例未做基因检测。鉴于患儿父亲皮肤和甲异常角化而没有多发性囊肿,结合患儿临床表现,故诊断为I型先天性厚甲症。由于皮肤角化异常,指(趾)甲异常增厚,故局部感觉减退、易发生浸渍糜烂,疼痛不适是最常见的症状。鉴于本病少见,有效的治疗报道不多,更无特异的治疗手段。治疗是对症性和经验性的,润滑剂和角质剥脱剂对轻症者有效[4]。应尽量减少反复的创伤和摩擦。
有研究表明维A酸类药物(异维A酸等)对本病有一定疗效[5]。外科切除严重变形甲(包括基质切除)后通常复发,因为PC中所有涉及的角蛋白(K6,K16和K17)都在甲床中有表达,K17在甲母质中也有表达,这也许能解释偶见的PC-II病例在基质切除后改善[1]。其他的治疗包括用水杨酸等软化角质层、止痛、局部理疗、畸形的矫正等综合疗法对改善患者生活质量有一定的帮助[6]。本例患儿因家庭原因,未口服维A酸类药物治疗,仅于渗出部位外用硼酸粉湿敷及抗生素软膏,干燥、皲裂部位外用尿素维E乳膏润滑保湿等对症治疗。目前仍在随访中。
PC是一种角蛋白基因突变所致的,以皮肤、甲增厚性营养不良为特点的常染色体显性遗传病,尚无特效预防和治疗方法。随着分子生物学技术的发展,目前可寄希望于未来的遗传咨询、胚胎植入前遗传性状筛选等基因治疗手段来减少本病的发生[7,8]。
猜你喜欢
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
火炸药学报(2021年2期)2021-05-06
电子产品世界(2021年4期)2021-02-09
家庭百事通·健康一点通(2020年3期)2020-08-20
东方教育(2017年14期)2017-09-25
山东工业技术(2017年17期)2017-09-13
海外文摘·文学版(2017年5期)2017-07-26
科学生活(2017年2期)2017-03-20
课程教育研究·学法教法研究(2016年1期)2016-03-17
