
Effect of mesenchymal stem cell in liver regeneration and clinical applications
2021-05-10 08:54SeongMinLeeSeungDukLeeSarahZiqiWangDevanandSarkarHannahLeeAamirKhanChandraBhatiAmitSharmaVinayKumaranDavidBrunoAdrianCotterellMarlonLevy
Hepatoma Research 2021年7期
Seong Min Lee, Seung Duk Lee, Sarah Ziqi Wang, Devanand Sarkar, Hannah M. Lee, Aamir Khan,Chandra Bhati, Amit Sharma, Vinay Kumaran, David Bruno, Adrian Cotterell, Marlon F. Levy
1School of Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA.
2Division of Transplant Surgery, Department of Surgery, Virginia Commonwealth University, Richmond, VA 23298, USA.
3Department of Human and Molecular Genetics, Virginia commonwealth University, School of Medicine, Richmond, VA 23298,USA.
4Division of Gastroenterology, Hepatology, and Nutrition, Virginia Commonwealth University, Richmond, VA 23298, USA.
#Authors contributed equally.
Abstract Liver disease accounts for approximately 2 million deaths per year worldwide with cirrhosis, viral hepatitis, and malignancy being the most common causes. Consequently, the regenerative capacity of the liver is a topic of extreme interest in the search for curative therapies to end-stage liver disease. Mesenchymal stem cells (MSCs)have emerged as a promising new therapy for hepatic regeneration. MSCs have multiple properties that make them an appropriate treatment option for liver disease including easy accessibility, targeted migration,immunomodulatory potential and antifibrotic/antioxidant effects. Additionally, MSCs have potential clinical applications in acellular therapy and tissue engineering. Liver regeneration with concurrent attenuation of liver injury makes MSCs a compelling therapeutic target in the setting of severe liver disease. This review outlines the mechanisms of MSC-driven liver regeneration and suggests potential clinical applications.
Keywords: Mesenchymal stem cell, liver regeneration, end-stage liver disease
INTRODUCTION
The liver is constantly subjected to noxious damage from both exogenous and endogenous toxins, thus requires a method to recover from injury. Normal liver regeneration is achieved primarily through proliferation of existing mature hepatocytes and biliary epithelial cells (BECs)[1]. Studies have demonstrated that regeneration of the liver following hepatectomies are characterized by phenotypic fidelity, meaning each cell is responsible for propagating its own cell type[2]. That is, hepatocytes make other hepatocytes, and the same applies to most other liver cell types including BECs and hepatic stellate cells (HSCs). Stem cells are not typically associated with physiologic liver proliferation, with the exception of Kupffer cells and liver sinusoidal endothelial cells (LSECs), both of which can be derived from bone marrow stem cells[3]. Of note,in the setting of impaired hepatocyte or BECs proliferation, the unaffected cell type can transdifferentiate into the impaired cell type and effectively function as facultative stem cells[4].
Despite the exceptional regenerative capacity of the liver, chronic injury can overwhelm the liver’s ability to regenerate and this leads to fibrosis. Liver fibrosis is a secondary wound healing process driven by myofibroblasts to degrade normal extracellular matrix (ECM) and accumulate excess connective tissue[5].The majority of myofibroblasts in liver fibrosis is derived from trans-differentiation of quiescent HSCs,which lead to activation of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs)[6]. A subset of myofibroblasts are derived from portal myofibroblasts and bone marrow (BM)-derived fibrocytes[7]. Portal myofibroblasts drive fibrogenesis exclusively in the biliary system, while the BM-derived fibrocytes minimally contribute to hepatic fibrosis[7]. Interestingly, fibrocytes share many phenotypic features with MSCs. Fibrocytes are BM-derived, collagen type 1 producing cells that produce ECM components and contribute to liver fibrosis. Fibrocytes appear to have regenerative properties and express surface markers like CD11b, CD14, CD34, CD45 and α-smooth muscle antibody (SMA) that are seen in cells of hematopoietic lineage[8]. However, fibrocytes lack the heterogeneity of MSCs and have unique proteomes that suggest BM-derived fibrocytes are distinct from MSCs[9]. Regardless of the source of myofibroblasts, they all express high levels of fibrillar collagen, TIMPs, and they are dominant contributors to liver fibrosis[5].
Currently, liver transplantation is the only definitive treatment for end-stage liver disease (ESLD).Fortunately, improvements in immunosuppressive drugs and surgical methods have improved transplantation outcomes and the global organ transplantation market is projected to grow significantly through the next few years[10]. The wide range of therapeutic potential of MSCs can further improve outcomes in ESLD as adjuvant or alternative therapy to liver transplantation. First, MSCs are pluripotent stem cells capable of differentiating into hepatocyte-like cells bothin vivoandin vitro[11,12]. Second, MSCs are readily accessible from multiple potential sources including adipose tissue, umbilical cord (UC),umbilical cord blood, peripheral blood, synovial membranes, muscle, dermis, and liver[13-17]. Importantly, the harvested MSCs maintain their pluripotent potential, robust proliferative ability, and capacity forex vivoexpansion[18]. Third, MSCs have the ability to migrate and engraft at sites of injured tissue[19]. Fourth, MSCs have immunosuppressive properties that allow for allogeneic transplantation. The immunosuppressive ability of MSCs also includes anti-fibrotic and antioxidant effects which can protect the liver from fibrosis and oxidative damage[20]. Lastly, MSCs produce extracellular vesicles (EV) that contain growth factors and cytokines that promote regeneration of impaired tissue such as liver parenchyma[20]. In this review, we will focus on the potential therapeutic mechanisms of MSCs and future studies that can help develop more effective treatments for ESLD [Table 1].
HOMING AND MIGRATION OF MSCS IN LIVER REGENERATION
A major criterion for effective stem cell therapy is the induction and engraftment of cells into the region ofdamage. The regenerative potential of MSCs in ESLD is largely reliant on the MSCs’ ability to migrate to the liver following administration. MSCs homing involves initial tethering by selectins, activation by cytokines,arrest by integrins, and extravascular migration towards chemokine gradients[31]. Tissue injury releases stress signals that attract MSCs to the site of damage[32]. Molecules expressed on MSC surfaces such as CXCR4, Eselectin, CD44, VLA-4, VCAM-1, and TIMP3 facilitate the subsequent adhesion, activation, and migration into damage tissue[33]. Unfortunately, MSC homing is inefficient and only a small percentage of cells reach the target tissue following systemic administration. In rat models of carbon tetrachloride (CCl4)-induced liver necrosis treated with green fluorescent protein (GFP)-labeled MSCs, the quantity of GFP-labeled MSCs detected in the liver was significantly less than what would be expected compared to the injected quantity[34].In vivostudies have shown that a large fraction of MSCs become trapped in the lung after intravenous injection[35]. However, there is evidence that pretreatment with vasodilators significantly increases MSC homing to the site of injury[35].

Table 1. Advantages and disadvantages among different cell types for treatment of liver disease
Multiple studies have demonstrated that the stromal cell-derived factor (SDF)-1a plays an important role in stem cell homing, chemotaxis, engraftment, proliferation, and survival[36-42]. SDF-1a is a chemotactic protein of the CXC family of proteins produced by bone marrow stromal cells. SDF-1a and its receptor chemokine CXC receptor 4 (CXCR4) are expressed in a variety of cells and tissues including immune cells, brain, heart,liver, kidney, lung, and spleen[43]. SDF-1a promotes the migration of stem cells to damaged tissue by binding to CXCR4 on stem cell membranes[40-42]. CXCR4 expression is endogenously regulated by tissue environmental factors such as cytokines, chemokines, stromal cells, adhesion molecules, tissue damage, and hypoxia[44]. For example, hypoxic conditions in the kidney appeared to increase CXCR4 expression of MSCs which enhanced functional recovery, accelerated mitogenic response, and reduced cell death[45]. This finding is further supported by rat models of myocardial infarction where increased expression of CXCR4 maximized the effect of SDF-1a to increase MSC migration and cardiac tissue regeneration[46].
SDF-1a is a potent chemoattractant for cells expressing CXCR4, and studies have demonstrated increased concentrations of SDF-1a in the liver following acute liver injury[47]. However, MSCs have low native levels of CXCR4 which could explain the poor mobilization of MSCs in the setting of liver disease[48].Furthermore, MSCs gradually downregulate the expression of CXCR4 after culturing and these cells lose the ability to migrate towards SDF-1a[49]. In a study of MSC stimulated regeneration of reduced size liver transplants, CXCR4 overexpressed rats exhibited enhanced MSC engraftment following MSC therapy with improved hepatocyte proliferation and increased survival compared to control[50]. Interestingly, the engrafted MSCs did not express markers of hepatocytes suggesting that these cells promoted regeneration of the remnant liver by a paracrine (relating to, promoted by, or being a substance secreted by a cell and acting on adjacent cells) mechanism. A subsequent study of genetically modified MSC with CXCR4 overexpression demonstrated similar results with increased migration and engrafted of MSC at the liver following acute liver failure[48]. This study also demonstrated significantly increased levels of hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF), again indicative of the presence of additional paracrine signaling that stimulates endogenous liver regeneration.
MSC DIFFERENTIATION INTO HEPATOCYTE-LIKE CELLS
MSCs are pluripotent stem cells with the potential to differentiate into cells of all three germ layers,including the endoderm, mesoderm, and ectoderm. In bothin vivoandin vitroexperiments, MSCs have demonstrated the ability to differentiate into hepatocyte-like cells with liver-specific morphology and function in the presence of cytokines and growth factors including HGF, fibroblast growth factor (FGF),oncostatin M (OsM), epidermal growth factor (EGF), leukemia inhibitory factor, and insulin-like growth factor (IGF)[51]. This pluripotent capacity is demonstrated by the presence of human hepatocyte markers such as albumin, α-fetoprotein (AFP), CK18, and CK19 in liver tissue of cirrhotic rats after human UCderived MSC administration[52]. These hepatocyte markers were not detected prior to MSC injection and there was no detection of rat-lineage hepatocyte albumin, AFP, CK18, or CK19, suggesting the transplanted human UC-derived MSCs were entirely responsible for differentiating into hepatocyte-like cells.
Although MSCs can be induced to differentiate in culture, an organ-specific microenvironment is the most suitable method for differentiation into a specific cell type[53]. Hepatic-differentiated cells are characterized by the expression of hepatocyte-specific genes and these genes are influenced by the microenvironmental conditions[54]. Studies have demonstrated that human UC-derived MSCs differentiated into hepatocyte-like cells more rapidly when in a fibrotic liver microenvironment, mimicked by rat fibrotic liver tissue extract[55].Allyl alcohol-treated rat liver models have demonstrated human MSCs ability to convert into functional hepatocyte-like cells when directly administered intra-hepatically to damaged rat liver[56]. Notably, liver function was restored within a week of transplantation through mechanisms suggestive of microenvironmental cues rather than cell fusion[56].
Zhanget al.[52]have demonstrated that MSCs do not directly differentiate into functional hepatocytes;instead, they first differentiate into BEC-like cells and subsequently differentiate to hepatocyte-like cells. In contrast, other studies indicate that trans-differentiation of MSCs is rare following MSC infusion in animal models[54]. For example, menstrual blood-derived MSCs were shown to inhibit HSC and liver fibrosis, thus improving liver function[54], but only a small fraction of the transplanted MSCs differentiated into functional hepatocyte-like cells[57]. Based on these findings, the current understanding is that MSCs exert their therapeutic effects through both direct cell differentiation and indirect paracrine signaling [Table 2].
SECRETION OF TROPHIC FACTORS FOR LOCAL AND SYSTEMIC SIGNALING
The therapeutic effects of transplanted MSCs were initially thought to be solely mediated by their migration to the site of injury, where integration and differentiation would take place. However, studies have shown that only a small proportion of MSCs have been observed to actually engraft and proliferate in the damaged tissue[58]. One proposed explanation is that MSCs elicit their therapeutic effects through the secretion of trophic factors. MSCs release a collection of trophic factors that signal for the regeneration of damaged tissues. These factors include growth factors, cytokines, and chemokines, which not only reduce the inflammation, apoptosis, and fibrosis of damaged tissues but also stimulate angiogenesis and tissue cell regeneration[58]. Local inflammatory cytokines, ligands of Toll-like receptors (TLRs), and hypoxic conditions all stimulate MSC migration to sites of cell damage. These conditions increase the production of MSCreleased growth factors and enhance the regenerative processes[59,60].

Table 2. Sources for MSC harvest, growth factors for differentiation, and markers of differentiation into hepatocyte-like cells
In the setting of liver fibrosis, MSC-secreted trophic factors can increase hepatocyte survival via antiapoptotic (stromal cell-derived factor 1, HGF, IGF-1, and VEGF), mitogenic [EGF, HGF, nerve growth factor (NGF), and transforming growth factor alpha (TGF-α)], and angiogenic effects (VEGF)[61-63]. Trophic factors lengthen the replicative cycle of injury and repair of both living and dying hepatocytes[63]. MSC transplantation was also associated with alterations in HGF and IGF-1 expression that correlated with reduced inflammation of fibrotic tissue during anti-apoptotic events[64,65]. HGF, EGF, and TGF-α are potent mitogens primarily associated with hepatocyte proliferation and VEGF enhances angiogenesis which is crucial for liver regeneration[66-69]. In addition to hepatocytes, hepatic progenitor cells, which are located in the Canals of Herring, can be differentiated into hepatocyte-like cells or biliary lineage cells following treatment with EGF or HGF, respectively[70]. Trophic factors, such as IL-10, HGF, NGF, TGF-β and tumor necrosis factor (TNF)-α regulate the proliferation of activated HSCs and decrease collagen synthesis in liver fibrosis[71].
Several studies on MSC-derived extracellular vesicles support the theory of paracrine signaling in MSCbased liver therapy. MSC-derived EVs contain MSC trophic factors that protected mouse liver against CCl4-induced injury by activating proliferative and regenerative responses[72]. A study demonstrated that EVs suppressed toxin-induced hepatocyte apoptosis by promoting the expression of anti-apoptotic protein B-cell lymphoma-extra large (Bcl-xL). EVs isolated from human UC-derived MSCs showed reduction in hepatic inflammation and collagen deposition of CCL4-induced fibrotic liver[73]. Additionally, there was an increase in liver aspartate transaminase (AST) activity, suggestive of functional liver recovery. MSC-derived EVs also demonstrated immunosuppressive properties associated with MSCs such as upregulation of T regulatory (Treg) cells and anti-inflammatory cytokines[74,75]. Further research on the exact mechanism of MSC-derived trophic factors is important for determining how MSCs exert their effects of liver regeneration and can subsequently lead to more targeted therapy for severe liver disease [Table 3].
MSC AND REGULATION OF IMMUNOLOGIC RESPONSE
MSCs exert an immunosuppressive effect through a multifaceted approach. MSCs have low expression of major histocompatibility complex (MHC)-II and costimulatory cell surface markers which is a major reason behind its allograft potential[76]. MSCs also directly interfere with immune response through direct cell-tocell interactions and secretion of soluble factors. For instance, MSCs can inhibit cell proliferation of T-, B-,natural killer (NK)-, and dendritic cells (DC) to induce cell division arrest anergy[77]. MSCs stop a wide range of innate and adaptive immune cell functions through the cytotoxicity of T and NK cells, B cell antibody secretion, inhibition of monocytes, DC antigen presentation, induction of apoptosis via programmed death (PD)-1, and upregulation of Tregs, especially in the setting of inflammation which activates MSCs[77].
MSCs secrete immunomodulatory factors, including nitric oxide (NO), prostaglandin E2 (PGE2),indoleamine 2,3-dioxygenase (IDO), human leukocyte antigen (HLA)-G, IL-6 and -10[77]. MSC induction of NO synthase (NOS) is a major mechanism for T cell suppression by MSCs, while IDO and TGF-β had morequestionable effects[78]. PGE2 exhibits multiple immunomodulatory effects, such as cell proliferation,apoptosis, tissue repair, angiogenesis, anti-inflammation, immune surveillance, and anti-cancer[79-81]. PGE2 release has significant anti-inflammatory properties, specifically by stimulating the synthesis of antiinflammatory cytokine IL-10 and by decreasing the synthesis of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and IL-12 from DCs. MSC-secreted PGE2 has additional antiinflammatory effects through suppression of T cells, macrophages, monocytes, and NK cells[82-84]. PGE2 also favors Th2 humoral immune responses over Th1 cellular immune responses by inhibition of IL-2 synthesis and increased proliferation of Treg cells[85]. MSCs further suppress B and T cell proliferation through IDO and HLA-G, which contribute to DCs maturation and NK cell cytotoxicity[86,87]. Secretion of IL-6 is another inhibitor of T-cell mediated immunity and directly disrupts the DC maturation process[88,89]. IL-6 also contributes to immune suppression by inhibiting apoptosis in lymphocytes and neutrophils. Lastly, MSCs can downregulate the immune response by suppressing macrophage polarization and generating tolerogenic DCs[78].
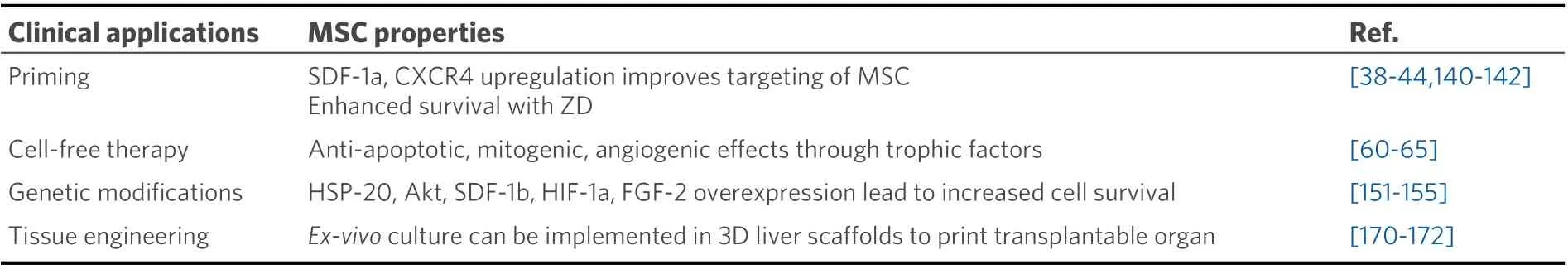
Table 3. Clinical application of MSCs in treatment of ESLD
In addition to immune suppression, MSCs can trigger active tissue regeneration and remodeling. MSCs secrete a variety of autocrine (a substance produced by a cell which stimulates its own secretion) and paracrine factors that support regenerative processes including angiogenesis, tissue repair, and regeneration[58]. MSC secreted trophic factors that facilitate the regeneration of specific tissues, including growth factors [brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor, EGF,FGF-2/-4/-7/-9/-17, HGF, IGF-1, NGF, and platelet-derived growth factor (PDGF)], cytokines (IFN-γ,TNF-α, and IL-1α/β, -2, -6, -8, -10, -12, and -13), chemokines (various CCLs and C-X-C motif ligands),antiapoptotic and angiogenic factors (VEGF), facilitating the regeneration of specific tissues[62]. The wide range of immunomodulatory effects of MSCs, from decreasing inflammatory responses to enhancing cellular repair demonstrates implications for treatment of severe liver disease as well as treatment for acute and chronic rejection following liver transplants.
ANTI-FIBROTIC AND ANTIOXIDANT PROPERTIES OF MSCS
Hepatic fibrosis can occur through a multitude of mechanisms including chronic liver injury from toxins(alcohol, drugs), viral infection, or metabolic imbalances. Fibrosis is a defining feature of chronic inflammation and is characterized by disruption of normal ECM architecture. HSCs are the key cells in hepatic fibrosis. HSCs can be activated from quiescent, vitamin A - storing cells to proliferative, α-SMApositive, myofibroblast-like cells with increased collagen synthesis[20]. MSCs have been reported in various animal models of heart, liver, kidney, lungs, pancreas, skin, peritoneum, and rectum to have anti-fibrotic activity[90].
MSCs are effective in treating fibrosis due to their antifibrotic and immunosuppressive properties. MSCs can upregulate the expression of MMP-2, -9, -13, and -14[60,91,92]which have recently been shown to reduce liver fibrosis by degrading ECM[60]. MSCs further augment this effect by downregulating the TIMP[93]. The balancing of MMPs and TIMP is associated with resolution of fibrosis[91]. MSCs also directly and indirectly suppress the activation and proliferation of HSCs, and thereby collagen synthesis. The direct interaction of MSCs with HSCs serves to suppress HSC proliferation by arresting them in G0/G1 phase through the inhibition of phosphorylation of extracellular signal-regulated kinase (ERK)1/2[59]. When MSCs are directly co-cultured with HSCs, MSCs suppress the α-SMA expression of HSCs, partially mediated by Notch pathway activation[94]. Indirectly, MSCs secrete trophic factors (IL-10 , HGF, TGF-B3, and TNF-α) that inhibit the proliferation of HSCs and decrease collagen synthesis[59,95], while HGF and NGF promote the apoptosis of HSCs[59,95,96]. Additionally, MSCs promote the expansion of NK cell population in liver and peripheral blood[97]. Subsequently, NK cells have been found to inhibit HSC activation by direct killing through IFN-γ and thus alleviate hepatic fibrosis[98]. This association was noted in rat models of cholestatic hepatic fibrosis where the intrahepatic NK cell levels were significantly decreased compared to control[97].Furthermore, T regulatory (Treg) cells exert pro-fibrotic effects by inhibiting NK cells[99]. However, this effect is inhibited by MSCs immunomodulation of T cell proliferation and thus contributes to the antifibrotic effects of MSCs. Collectively, MSCs inhibit HSCs, and collagen synthesis, suppress overactive immune reactions and ultimately restrain fibrosis[59].
Oxidative stress from reactive oxygen species (ROS) is a common mechanism of liver injury leading to liver fibrosis, cirrhosis, viral hepatitis, hepatocellular carcinoma (HCC), and others[100-104]. Several studies have suggested that MSCs mediate strong antioxidant effects in various animal models[105-108]. Carbon tetrachloride (CCl4) and thioacetamide (TAA) are the most commonly used toxins to simulate oxidative liver damage[109,110]. These toxins cause hepatocyte damage through lipid peroxidation, and alkylation of proteins, nucleic acids, and lipids[105,106,111]leading to inflammation, hepatocellular damage, and fibrosis.Notably, the small amount of physiologic ROS produced by cellular respiration is necessary in cell signaling and homeostasis[112,113]. MSCs have demonstrated the capacity to alleviate CCl4- and TAA-induced oxidative stress bothin vitroandin vivo[105,107]. MSCs upregulate the expression of superoxide dismutase and induction of AREs, thus enhancing antioxidant and cytoprotective activity to reduce hepatocyte apoptosis[105,107]. MSCs’antioxidant effects have also been observed extra-hepatically in diseases such as dextran sulfate sodiuminduced colitis and neurodegenerative diseases (e.g., Friedreich’s ataxia)[114]. The antioxidant effects of MSCs, combined with their immunomodulatory effects, are promising properties for the development of therapies to treat liver disease [Figure 1].
CLINICAL APPLICATION OF MSC IN LIVER REGENERATION
MSC-based cell therapy has multiple advantages over conventional liver transplantation or hepatectomies.MSCs are relatively easy to harvest from multiple different sources and they maintain their pluripotency following injection. A study by Kharazihaet al.[115]demonstrated that intravenous infusion of autologous bone marrow (BM)-derived MSCs among patients with decompensated cirrhosis resulted in statistically significant improvements in liver function, as measured by model for ESLD score (17.9-10.7), international normalized ratio (INR) (1.9-1.4), serum creatinine (114-80), serum albumin (30-33), and bilirubin (46-41)with no adverse events noted. Similarly, phase II clinical trials have shown similar results in alcoholic cirrhosis with histologic and quantitative improvement of hepatic fibrosis following MSC injection via hepatic artery[116,117]. The feasibility and safety of MSC transplantation is further supported by phase I studies showing improvements in liver function with MSC injection in liver disease of various causes with no noted adverse effects[118]. According to the National Institute of Health (NIH), there are currently 55 active clinical trials involving MSC-based therapy for liver disease of various etiology including cirrhosis, acute liver failure, and hepatitis[119]. The MSCs used in these trials are derived from various sources including bone marrow, umbilical cord, adipose tissue, and menstrual blood. Most studies are using allogenic cell transplantation through peripheral veins, but there are cases of hepatic artery or portal vein injections. In multiple phase II studies of chronic hepatitis B and C, treatment with HSCvs. standard supportive treatment showed significant improvement in Child-Pugh and MELD scores[27,120,121]. In chronic hepatitis B patients, MSC infusion resulted in improved ascites and fibrosis markers as well as MELD scores[122]. MSC also appeared to be more effective when administered via portal vein than peripherally[120]. There was no significant difference in improvements between direct MSC injection compared to pre-differentiation of MSC to hepatocyte-like cells before transplantation[27][Table 4].
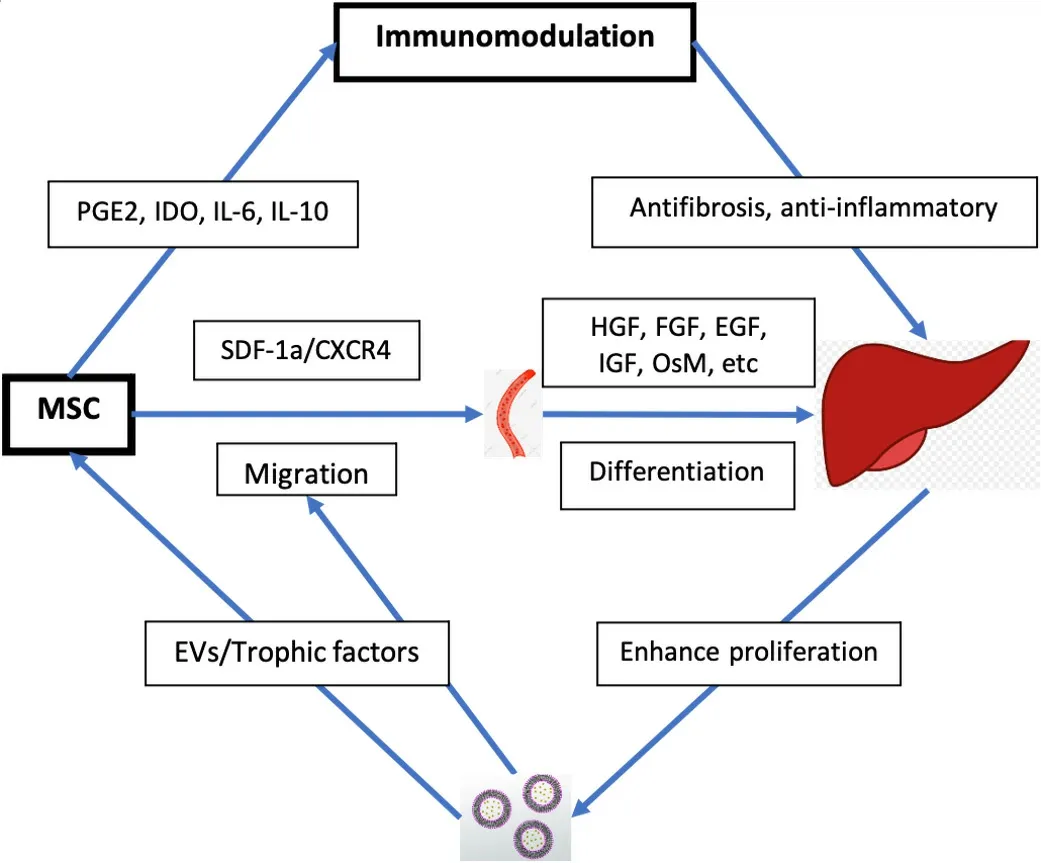
Figure 1. Therapeutic mechanisms of MSC in liver disease. MSC: Mesenchymal stem cell; PGE2: prostaglandin E2; IDO: dioxygenase;SDF-1a: stromal cell-derived factor-1a; CXCR4: CXC receptor 4; HGF: hepatocyte growth factor; FGF: fibroblast growth factor; EGF:epidermal growth factor; IGF: insulin-like growth factor; OsM: oncostatin M; EVs: extracellular vesicles.
ADMINISTRATION AND DOSING OF MSC-BASED THERAPY
MSCs were first isolated from bone marrow (BM) in 1970[127]. Since then, MSCs have been found in various tissues including adipose tissue, UC (Umbilical Cord blood), peripheral blood, synovial membranes,muscle, dermis, liver, and many others[13-17]. There is no established standard for procurement of MSCs for the treatment of liver disease, but they are most often harvested from the UC or BM because these are the most well studied[128]. Between BM-derived MSCs and UC-derived MSCs, UC-derived MSCs are the preferred cell type for multiple reasons. First, MSC-based treatment requires large amounts of MSCs and the UC provides significantly larger quantities of MSCs compared to BM[129,130]. Second, UC-derived MSCs can be isolated without the invasive procedures required for BM harvesting for MSCs[131]. Third, UC-derived MSCs are at an earlier phase of organic development compared to BM-derived MSCs, thus showing higher self-renewal and differentiation capacity[132]. Lastly, UC-derived MSCs have demonstrated lower immunogenicity than that of BM-derived MSCs suggesting superior allogeneic transplantation capability[133,134].
Clinical trials have demonstrated various routes of administration for MSC-based liver therapy. The most common methods of administration were peripheral intravenous injection, followed by portal vein, hepatic artery, and intrasplenic injections[128]. Peripheral intravenous injection has an obvious advantage ofconvenience; however, animal models have demonstrated that 60% of the injected MSCs accumulated in the lungs and never reached the liver[135]. Similar results were seen with intravenously injected green fluorescent protein (GFP)-labeled MSCs in mice models. The GFP-labeled MSCs were found in high concentrations in the lung, but not the liver[136]. Interestingly, studies of miniature pigs with acute liver failure demonstrated restoration of hepatic function following intraportal injection of MSCs, but this effect was not seen in peripheral vein administration[137,138]. In a study comparing the administration of BM-derived MSCs through portal vein and intrasplenic injection for liver failure, results showed portal vein administration was more effective in improving model for end-stage liver disease (MELD) score compared to intrasplenic injection[120]. However, this difference was not seen after the first month of treatment. Based on these results,intraportal injection appears to be the best method; however, further research on the long-term recovery of hepatic function and occurrence of complications such as infection or venous thrombosis could help optimize the administration of MSC-based liver therapy.
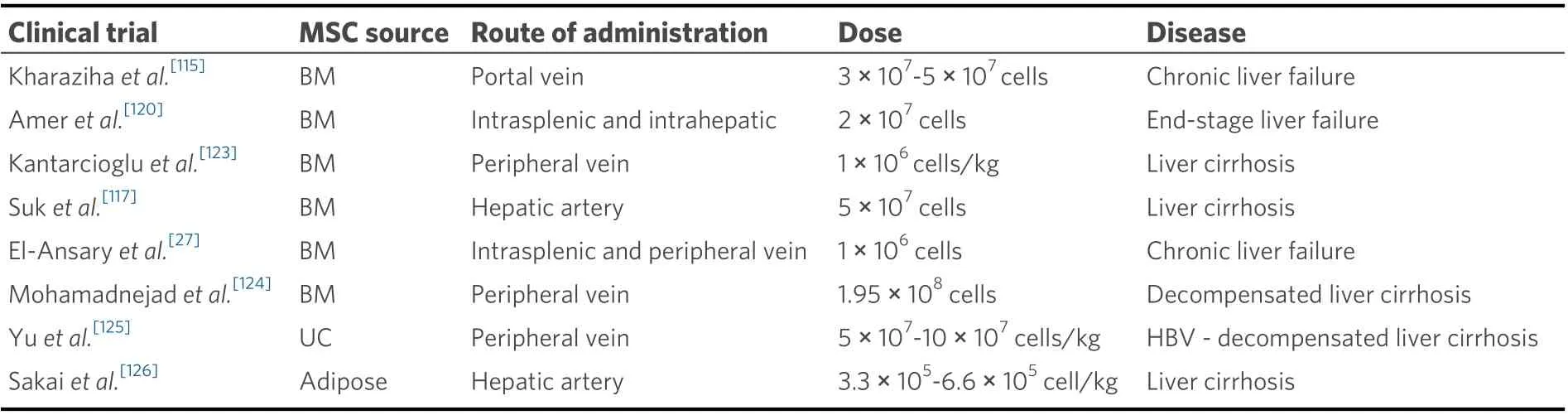
Table 4. Summary of clinical trials with MSC for ESLD
The most effective dosing and frequency for MSC-based treatment of liver disease are unclear. Most clinical trials used a body-weight based dosing within the range (0.5 × 106-3 × 106cells/kg) for a single dose, while some studies used total MSC quantity (1 × 107-20 × 107cells)[128]. Studies have shown significant improvement of liver fibrosis with doses as low as 1 × 107MSCs[139], and other studies demonstrated no improvement of liver fibrosis at a substantially higher dose of 2 × 108MSCs[124]. Most studies administered one-time doses, but in a study comparing a single dose to two doses a month apart, there was no significant difference in improvement of liver cirrhosis[117]. Further studies comparing a wider range of doses and frequencies are critical to generating an effective therapeutic dose.
MSC PRIMING
When MSCs are introduced to damaged liver, they can express various immune regulatory factors that can promote liver regeneration; examples of these include NO, PGE2, IDO, IL-6, IL-10, and HLA-G. However,depending on the concentration of inflammatory cytokines such as IFN-γ, TNF-α, and IL-1β, MSCs may lead to increased myofibroblast activity and worsen hepatic fibrosis[140,141].In vitroMSC priming is one way to enhance the therapeutic effects, while limiting the unwanted pro-fibrotic properties. IFN-γ primed MSCs induce IDO expression to inhibit the activation of T and NK cells[142]. The anti-inflammatory effect was increased when MSCs were primed with both IFN-γ and TNF-α[143]. These cells were less susceptible to NK cell-mediated killing and had enhanced immunosuppression[144]. Additionally, MSC-based therapy has low efficiency partly due to poor cell survival following transplantation and priming can increase cell survival[145]. Zeaxanthin dipalmitate (ZD) is a molecule that has been demonstrated to enhance the survivability of MSCs[146]. Pretreatment with ZD dramatically improved cell survival by suppressing apoptosis, inflammation, and ROS production in adipose-derived MSCs[146]. Further investigation is needed to determine whether the beneficial effects of priming MSCs prior to transplantation are maintained longterm after MSC differentiation.
MSC-BASED CELL-FREE THERAPY
MSCs secrete small extracellular vesicles, also known as exosomes, which contain proteins and factors that can exert specific actions on local and distant targets. MSC-derived EVs have been shown to reduce injury following myocardial ischemia, acute kidney injury, and acute liver failure but its mechanism of hepatoprotective effects is unclear[73]. In CCl4 mice models, the administration of MSC-derived exosomes led to inhibition of collagen production, which reduced hepatic inflammation and fibrosis[73]. MSC-derived exosomes have shown therapeutic effects in setting of acute liver injury in various mouse models.Specifically they have been demonstrated to increase hepatocyte proliferation, upregulate liver regenerative genes, and increase production of proliferative proteins such as cyclin D1, Bcl-xL, and signal transducer and activator of transcription (STAT)-3[72]. Intra-tumor administration of MSC-derived exosomesin vivohas been shown to significantly reduce tumor growth[147].
Direct transplantation of MSCs has associated risks including tumorigenesis and fibrogenic potential that can raise concerns with MSC cell-based therapy. Given that multiple studies have suggested the therapeutic effects of MSCs are exerted through MSC-derived exosomes, an acellular approach may provide similar benefits of cell-based therapy with fewer risks. MSC-derived exosomes also have great potential to be an intracellular drug delivery vehicle. MSC exosome-shuttle therapy has been used in regenerative medicine following ischemic cardiac injury[42]. MSC-derived exosomes have also been used to deliver cytokines such as IFN-β, IFN-α2b, tumor necrosis factor-related apoptosis inducing ligand (TRAIL), and IL-12 for liver cancer therapy[148,149,150,151-153]. CXCR4-enriched exosomes have shown beneficial effects due to upregulation of protein kinase B (Akt) signaling pathway that promotes cell survival and angiogenesis[154,155]. Currently,studies involving MSC-derived exosome-based therapy for liver disease are limited in number. Additional research on the potential application of MSC-derived exosomes could provide a novel approach for treatment of cirrhosis, hepatocellular carcinoma, and hepatitis.
GENETIC MODIFICATION OF MSCS
MSC-based therapy for liver disease is limited by several factors such as low rates of cell survival, poor engraftment, and inefficient homing mechanisms. Fortunately, these limitations can be overcome through genetic modifications. A variety of pro-survival genes have been inserted into MSCs to prolong their survival in target organs. For example, overexpression of genes for heat shock protein (HSP)-20, (Akt),SDF-1β, hypoxia-inducible factor (HIF)-1α, FGF-2, all increased cellular survival by providing protection from oxidative stress, ischemia, hypoxia, and apoptosis[156-160]. Additionally, SDF-1 and CXCR4 engineered MSCs exhibited efficient homing and engraftment leading to greater regeneration of multiple organs including the liver[50,161,162]. Decorin (DCN), an important component in ECM, has demonstrated antifibrotic effects in liver by facilitating ECM degradation. DCN-modified MSCs exerted strong protective effects against hepatic fibrosis by suppressing TGF-β/Smad signaling[163]. Transplantation of urokinase-type plasminogen activator (uPA) in the MSC gene showed lower expression of α-SMA, TGF-β1, and collagen types I/III with increased expression of MMP-2, -3, -3, HGF, and proliferating cell nuclear antigen. The overall effect was suppression of hepatic fibrosis and improvement in liver function[164]. High levels of IL-10 secretion from MSCs were associated with improved liver regeneration. IL-10 gene transfer into MSCs used as novel treatment in rat liver fibrosis models demonstrated suppressed HSCs, improved liver histopathology, and increased liver function[165]. These studies suggest that genetic modification of MSCs has the potential to improve survival, targeting, and pro-regenerative capacities of MSCs. However, the potential tumorigenic effects of MSCs should be considered and safety of treatment should be evaluated before genetic modifications are implemented in treating liver disease.
MSC-BASED TISSUE ENGINEERING
A recently developed strategy to improve liver regeneration is through tissue engineering of 3D scaffolds.MSCs exhibit improved cell proliferation and hepatic differentiation into characteristic mature hepatocytes when cultured in 3D biomatrix scaffold compared to in 2D substrates[166].In vitrosynthesis of liver scaffold can be generated from natural[167]and synthetic[168]materials, fluid flow[169], 3D culture[168,169], or 3D bioprinting[170-172]. 3D spheroid MSC cultures have been shown to improve the differentiation efficiency of MSCs and to enhance therapeutic potential[173,174]. Spheroid cultures of MSCs increased the expression of antifibrotic factors and had greater hepatoprotective effects when compared to 2D cultured MSCs.Uygunet al.[175]demonstrated that a 3D architecture of decellularized liver could be re-cellularizedin vitroby MSCs and maintain viability after transplantation. This implies a transplantable liver can be synthesizedin vitro. Similar success was seen with hepatobiliary organoids that could survivein vivowhile maintaining functionality of hepatic and biliary structures[176,177]. Several additional studies have had success within vitrogeneration of liver organoid and subsequent transplantation with good functionalityin vivo. These studies suggest that transplantation of liver organoids in the setting of acute liver failure may prolong survival[178,179].More research would be required to directly compare the benefits of liver organoid therapyvs. MSC-based cell therapy [Figure 2].
RISKS OF MSC THERAPY
Despite several clinical trials that have demonstrated the safety and efficacy of MSCs in liver diseases, it is important to note MSC therapy is not without risks. As discussed previously, MSCs have the ability to migrate to the liver and promote regeneration through immunosuppression, antifibrotic, and antioxidant effects. However, there are circumstances in which MSC may cause harm due to their fibrogenic potential.MSCs have fibrogenic potential when cultured in hepatogenic differentiation medium containing HGF,FGF-4, and OsM. When these MSCs were transplanted into the liver of NOD/SCID mice undergoing partial hepatectomy, they expressed α-SMA, a marker for myofibroblast differentiation[140]. Additionally,transplanted MSCs in normal and acutely injured NOD/SCID mice models showed lower engraftment rates compared to chronically injured mice and greater number of MSCs in the acute liver injury models exhibited a myofibroblast-like morphology[141]. These results highlight the need for further evaluation of the potential contraindications of MSC therapy in the treatment of hepatic fibrosis.
The potential of MSCs for anti-tumor therapy stems from their ability to migrate and incorporate into tumor stroma[180]. There is conflicting evidence as to whether MSCs suppress tumor growth or contribute to tumor growth, through promotion of tumor-associated fibrosis, immunosuppression, angiogenesis, and metastasis[181]. Certainly, this poses a risk in MSC-based therapy for liver disease. Tumor cells secrete transforming growth factor (TGF)-β, which induce MSCs to preferentially differentiate into pro-fibrotic cells expressing α-SMA, tenascin C, and fibroblast surface protein[181]. Additionally, the tumor microenvironment increases the secretion of growth-stimulating factors such as CCL5/RANTES and stromal cell-derived factor-1 (SDF-1)[180]. MSCs can promote proliferation of tumor cells by differentiation into carcinoma-associated fibroblasts or tumor-associated fibroblasts, which express α-SMA, induce neovascularization and express tumor-stimulating factors[182,183]. Furthermore, MSCs exert antiapoptotic effects through factors including VEGF, FGF-2, HGF, BDNF, SDF-1α, IGF-1, and TGF-β that can support tumor growth. This effect is compounded by hypoxia in the tumor microenvironment which stimulates increased production of pro-survival factors[184-187]. Genetic modification and pretreatment of MSCs may be effective in decreasing the fibrogenic and tumorigenic potential of MSCs and increasing the safety profile of MSC-based therapy.
CONCLUSION
Currently, liver transplantation is the only definitive treatment for end-stage liver disease. Although the global organ transplantation is expanding and transplantation outcomes are improving through advancements in immunosuppressive drugs and surgical techniques, there is still significant room for progress. MSC-based therapy may offer a chance for more patients to receive a potentially curative treatment. MSCs can migrate to damaged liver tissue, differentiate into hepatocyte-like cells, reduce inflammation, decrease fibrosis, and have antioxidant effects. Multiple clinical trials have verified the safety and efficacy of MSC therapy for severe liver diseases of various etiologies including cirrhosis, liver failure,and post-transplant complications. Further studies with larger samples and blinded trials need to be conducted before MSCs can become an accepted alternative to liver transplant. Additionally, more clinical trials are necessary for optimizing MSC injection routes, dosing, frequency, and mechanism of delivery.Future studies on MSC priming, homing mechanism, MSC derived exosome therapy, genetic modifications,and tissue engineering could greatly improve MSC therapy and may create novel approaches to treating end-stage liver disease.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and writing of this paper and performed extensive literature review and interpretations: Lee SM, Lee SD, Wang SZ, Levy MF
Made contributions by providing comments and feedback throughout the writing of this paper: Sarkar D,Lee HM, Khan A, Bhati C, Sharma A, Kumaran V, Bruno D, Cotterell A
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.

- Hepatoma Research的其它文章
- A practical approach to pediatric liver transplantation in hepatoblastoma and hepatocellular carcinoma
- 3D Organoid modelling of hepatoblast-like and mesenchymal-like hepatocellular carcinoma cell lines
- BOOST: a phase 3 triaI of sorafenib vs. best supportive care in first Iine treatment of hepatoceIIuIar carcinoma in patients with deteriorated Iiver function
- Prognostic factors associated with survival in patients with hepatocellular carcinoma undergoing transarterial chemoembolisation: an Australian multicenter cohort study
- AUTHOR INSTRUCTIONS