
BOOST: a phase 3 triaI of sorafenib vs. best supportive care in first Iine treatment of hepatoceIIuIar carcinoma in patients with deteriorated Iiver function
2021-05-10 08:54GennaroDanieIeCIorindaSchettinoLauraArenareDomenicoBiIanciaFabioFarinatiPieraFedericoStefanoTamberiGinoCriveIIariSandroBarniRaffaeIIaTortoraFrancescoIzzoAntonioFrassoIdati0LuigiCavannaCIaudiaMucciariniLuigiBoIondiAngeIoDinot
Hepatoma Research 2021年7期
Gennaro DanieIe, CIorinda Schettino, Laura Arenare, Domenico BiIancia, Fabio Farinati, Piera Federico, Stefano Tamberi, Gino CriveIIari, Sandro Barni, RaffaeIIa Tortora, Francesco Izzo, Antonio FrassoIdati0, Luigi Cavanna, CIaudia Mucciarini, Luigi BoIondi, AngeIo Dinota, FiIippo PeIizzaro,Maria CarmeIa PicciriIIo, Piera GargiuIo, Massimo Di Maio,b, Ciro GaIIo4, Francesco Perrone,#, Bruno DanieIe,#
1Unità Sperimentazioni Cliniche, Istituto Nazionale Tumori, IRCCS, Fondazione G. Pascale, Napoli 80131, Italy.
2Oncologia Medica, Azienda Ospedaliera S. Carlo, Potenza 80131, Italy.
3Unità di Gastroenterologia, Dipartimento di Scienze Chirurgiche, Oncologiche e Gastroenterologiche, Università di Padova,Padova 35121, Italy.
4Oncologia, Azienda Ospedaliera G. Rummo, Benevento 82100, Italy.
5Oncologia Medica, Ospedale Civile degli Infermi, Faenza (RA) 48018, Italy.
6Oncologia Medica 1, Istituto Oncologico Veneto, Dipartimento di Oncologia Clinica e Sperimentale, Padova 35128, Italy.
7Oncologia Medica, Azienda Ospedaliera Treviglio - Caravaggio, Treviglio (BG) 24047, Italy.
8UOC Epatologia, Dipartimento dei Trapianti, AORN A. Cardarelli, Napoli 80131, Italy.
9Chirurgia Epato-biliare, Istituto Nazionale Tumori, IRCCS Fondazione G. Pascale, Napoli 80131, Italy.
10Oncologia Clinica, Azienda Ospedaliera Universitaria Arcispedale Sant' Anna, Ferrara 44124, Italy.
11Oncologia Medica ed Ematologia, USL di Piacenza, Ospedale Guglielmo da Saliceto, Piacenza 29121, Italy.
12UOC Medicina Oncologica, Ospedale Ramazzini, Carpi (MO) 41012, Italy.
13Medicina Interna, Policlinico S. Orsola-Malpighi, Bologna 40138, Italy.
14Statistica Medica, Università della Campania Luigi Vanvitelli, Napoli 81055, Italy.
a(Present addresses) Fondazione Policlinico Universitario A. Gemelli, IRCCS, Univer-sita' Cattolica, Roma, Italy.
b(Present addresses) Dipartimento di Oncologia, Università degli Studi di Torino - Oncologia, A. O. Ordine Mauriziano, Ospedale Umberto I, Torino, Italy.
c(Present addresses) Oncologia, Ospedale del Mare, Napoli, Italy.
★Co-first author; #co last authors.
Abstract Aim: Only patients with good liver function {[Child-Pugh (CP)] A class} were eligible for trials testing sorafenib as first-line treatment of hepatocellular carcinoma (HCC); nevertheless, the drug was authorized without restrictions based on liver function. Therefore, we planned to test sorafenib efficacy and safety in patients with HCC and deteriorated liver function (CP-B).Methods: This was an open-label, multicenter, randomized phase 3 trial. Patients with HCC, no previous systemic therapy, and CP-B score 7-9 were assigned 1:1 to best supportive care alone (control arm) or with standard dose sorafenib (experimental arm). Overall survival (OS) was the primary endpoint. To detect a 0.70 HR of death, with 80% power, and two-tailed α error 0.05, 234 events were required. The study closed prematurely because of slow accrual. Descriptive analyses are reported.Results: From 2012 to 2017, 13 Italian centers randomized 35 patients. In total, 28 deaths were recorded, 12 without and 16 with sorafenib; median OS was 4.9 (95%CI: 1.2-5.6) and 3.5 months (95%CI: 1.3-5.3), respectively.At least one severe adverse event was reported in 2/15 (13.3%) without and 9/17 (52.9%) patients with sorafenib.Conclusions: This trial failed its planned enrolment goal, showing the difficulty in performing clinical trials with drugs already registered with a label broader than what available evidence supports.
Keywords: Hepatocellular carcinoma, Child-Pugh B class, sorafenib
INTRODUCTION
In 2018, hepatocellular carcinoma (HCC) represented the 6th most common human cancer (over 840,000 new cases) and the fourth most common cause of cancer-related death (780,000 estimated deaths),according to the International Agency for Research on Cancer database[1]. Estimated ranking of incidence and mortality in 2019 in the United States suggests that incidence is going to decrease as compared to other types of cancer, but mortality remains significant[2]. Prognosis depends on both the tumor characteristics and the liver failure due to concomitant cirrhosis; thus, Child-Pugh score and other markers of liver function are included in several HCC staging systems[3-5].
In patients with advanced untreated HCC, the prognosis is extremely poor, yielding a median survival of 4-7 months[6].
Sorafenib is an oral multi-kinase inhibitor of vascular endothelial growth factor receptors 1, 2, and 3, the platelet-derived growth factor receptor, and the RAF pathways[7]. In two randomized phase 3 trials (SHARP and Asia-Pacific), it prolonged overall survival and time to progression compared to placebo in patients with advanced HCC who had not received prior systemic therapy[8,9].
In both trials, only patients with good liver function (Child-Pugh A) were eligible and few Child-Pugh B cases (20 and 6 patients in SHARP and Asia Pacific, respectively) were enrolled as protocol violations.Nevertheless, the Food and Drug Administration and European Medicines Agency approved sorafenib for the treatment of patients with advanced HCC regardless of liver function. However, the cost-benefit ratio is unknown in patients with highly compromised liver function, and sorafenib is reimbursed only for Child-Pugh A patients in several countries (Australia, Belgium, Canada, France, Italy, South Korea, Taiwan, and Switzerland)[10].
In 2011, we planned the BOOST (B Child-Pugh HCC patients - Optimization Of Sorafenib Treatment)randomized phase 3 trial to assess the efficacy and safety of sorafenib plus best supportive care (BSC)vs.BSC alone in Child-Pugh B advanced HCC patients.
PATIENTS AND METHODS
Study management and design
BOOST (NCT:01405573, EudraCT number: 2009-013870-42) was an open-label, multicenter, randomized phase 3 trial promoted by the Istituto Nazionale per lo Studio e la Cura dei Tumori - IRCCS Fondazione G.Pascale, Napoli, Italy. The study was supported by the Italian Drug Agency (AIFA) in May 2011 (code FARM84SA2X) for trial coordination activities. A request to the pharmaceutical company for gratuitous drug supply was unsuccessful. Therefore, only 15 centers, willing to pay for the experimental drug, accepted to participate, but the enrolment was quite null. In August 2014, AIFA agreed on further funding to buy the experimental drug. The number of centers willing to participate increased to 36, but only 13 actually enrolled at least one patient. Figure 1 summarizes time dynamics of the trial.
Patients were randomly assigned, in a 1:1 ratio, to receive BSC alone (control arm) or combined with sorafenib (experimental arm) until disease progression, occurrence of unacceptable toxicity, or progression of the underlying cirrhosis.
Participants provided written informed consent before any study procedures.
EIigibiIity criteria
Eligible patients had liver function classified as Child-Pugh B and advanced HCC (according to the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver criteria)[11], were not eligible for loco-regional treatment, and had not received previous systemic treatment. Other inclusion criteria were age ≥ 18 years, Eastern Cooperative Oncology Group (ECOG)performance status (PS) score 0-2, life expectancy longer than 2 months, adequate hematologic (platelet count ≥ 60.0 × 109/L; hemoglobin > 9 g/dL) and renal function (serum creatinine < 1.5 times the upper limit of normal range), and signed informed consent.
Key exclusion criteria were the presence of any unstable systemic disease or medical contraindication to sorafenib and any grade encephalopathy or gastrointestinal hemorrhage within 30 days before the randomization.
Treatment and study procedures
Sorafenib, at the starting dose of 800 mg (two 200 mg tablets every 12 h), was assumed orally on a continuous daily basis; for convenience, the treatment period was divided into 4-week cycles. Based on the occurrence and the severity of side effects (diarrhea, skin toxicity, hematologic, or other non-hematologic adverse events) or Child-Pugh deterioration, sorafenib was allowed to be stepwise reduced to 400 mg daily(200 mg bid) or 400 mg every other day. After such reductions, in case of persistent toxicity, treatment had to be definitively stopped.
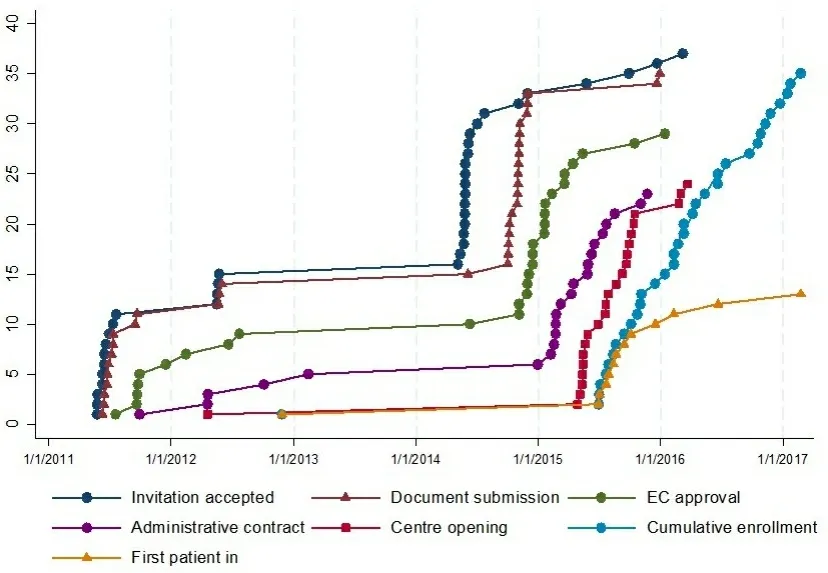
Figure 1. Time dynamics of trial conduction.
Randomization was performed centrally, at the Clinical Trials Unit of the Istituto Nazionale per lo Studio e la Cura dei Tumori - IRCCS Fondazione G. Pascale, Napoli, Italy, through a web-based minimization procedure, with Child-Pugh score (7vs. 8vs. 9), Cancer Liver Italian Program (CLIP) score (1vs. 2-3vs. 4-5), and age (< 70vs. ≥ 70) as strata.
Endpoints
The primary end point of the study was overall survival (OS), defined as the time between the date of randomization and the date of death from any cause.
Secondary endpoints included toxicity, quality of life (QoL), and progression-free survival (PFS).
Toxicity was evaluated in both arms at baseline and every four weeks until disease progression. Adverse events were coded according to Common Terminology Criteria for Adverse Events version 4.03. Toxicity was described for each item as the worst grade suffered by the patient at any time during the treatment.
QoL was measured with the European Organization for Research and Treatment of Cancer (EORTC) QLQC30 questionnaire and the HCC specific module (EORTC QLQ-HCC 18)[12,13]at baseline and every four weeks, until 24 weeks, in both treatment arms.Baseline tumor measurement, with abdominal and pelvic computed tomography scan or magnetic resonance, was planned for all patients, before the randomization, and in the sorafenib arm only, every eight weeks during treatment, until disease progression. PFS, defined as the time between the date of randomization and the date of disease progression [according to the modified Response Evaluation Criteria in Solid Tumors (RECIST 1.1)][14]or death, whichever occurred first, was described only in the sorafenib arm. For this purpose, patients who did not progress or die were censored at the date of the last available information on vital status.
StatisticaI anaIysis
Sample size calculation was based on an expected median survival of 4.5 months in the control arm and an auspicated 6.5-month median survival in the sorafenib arm, corresponding to a hazard ratio (HR) of death of 0.70. With a two-tailed alpha error of 0.05, and 80% power, 234 events were required, and it was planned to enroll 320 patients in two years (EAST 3.1, Cytel Software, Cambridge, MA, USA).
In March 2017, the study was stopped due to slow enrolment, with 35 patients randomized. Due to low study power, only descriptive analyses are reported.
Median follow-up was calculated according to the Schemper’s reverse Kaplan-Meier technique[15].
Survival curves were drawn with the Kaplan-Meier product-limit method, and the HR of death was calculated with the Cox proportional hazard model.
Toxicity was described for all the patients with available toxicity information. Side-effects were grouped either as any grade (Grade ≥ 1) or as severe (Grade ≥ 3).
Only baseline QoL was descripted due to the high number of missing QoL data through the follow up.
Analyses were performed using STATA MP 14.1 (StataCorp LP, College Station, TX, USA).
RESULTS
Patients’ characteristics
From 27 November 2012 to 2 February 2017, 13 Italian centers enrolled 35 patients (18 were assigned to the experimental and 17 to the control arm). One patient in the control arm withdrew consent immediately after randomization [Figure 2].
Notwithstanding the small number of patients, the baseline characteristics were well balanced between study arms [Table 1]. Median age was 64.8 years [interquartile range (IQR): 59.2-71.3 years]; 85.3% of patients were male, and 73.0% had an ECOG PS 0-1. Child-Pugh scores 7, 8, and 9 were similarly represented in the study population; only few patients (8.8%) were in the lowest CLIP score category, the majority of the patients (64.7%) being in the CLIP 2-3 category. Chronic hepatitis C virus infection was the predominant cause of liver disease (67.7% of the cases), followed by alcohol consumption, and HBV infection. Overall, 16 patients (47.1%) had been previously treated with locoregional therapy.
Treatment compIiance
Data on compliance to sorafenib are missing for one patient. The median duration of sorafenib treatment was 28 days (IQR: 20-60 days). The profile of dosing per day across the first two cycles is reported in Figure 3. There were three violations regarding the sorafenib initial dose, with a starting dose of 400 mgdaily rather than 800 mg. Four patients required a dose reduction during the treatment. At the time of the analysis, all patients had discontinued sorafenib, 11 of them after one cycle. Reasons for treatment discontinuation were progression of HCC (6 cases), toxicity (5 cases), patient refusal (3 cases, 2 of whom with diarrhea), and worsening from Child-Pugh B to C (2 cases). Adverse events leading to treatment
discontinuation were bilirubin increase (2 cases), fatigue (2 cases), and abdominal pain, nausea, vomiting,skin rash, and anemia (1 case each); multiple adverse events were associated in two patients.

Table 1. Characteristics of patients according to study arm
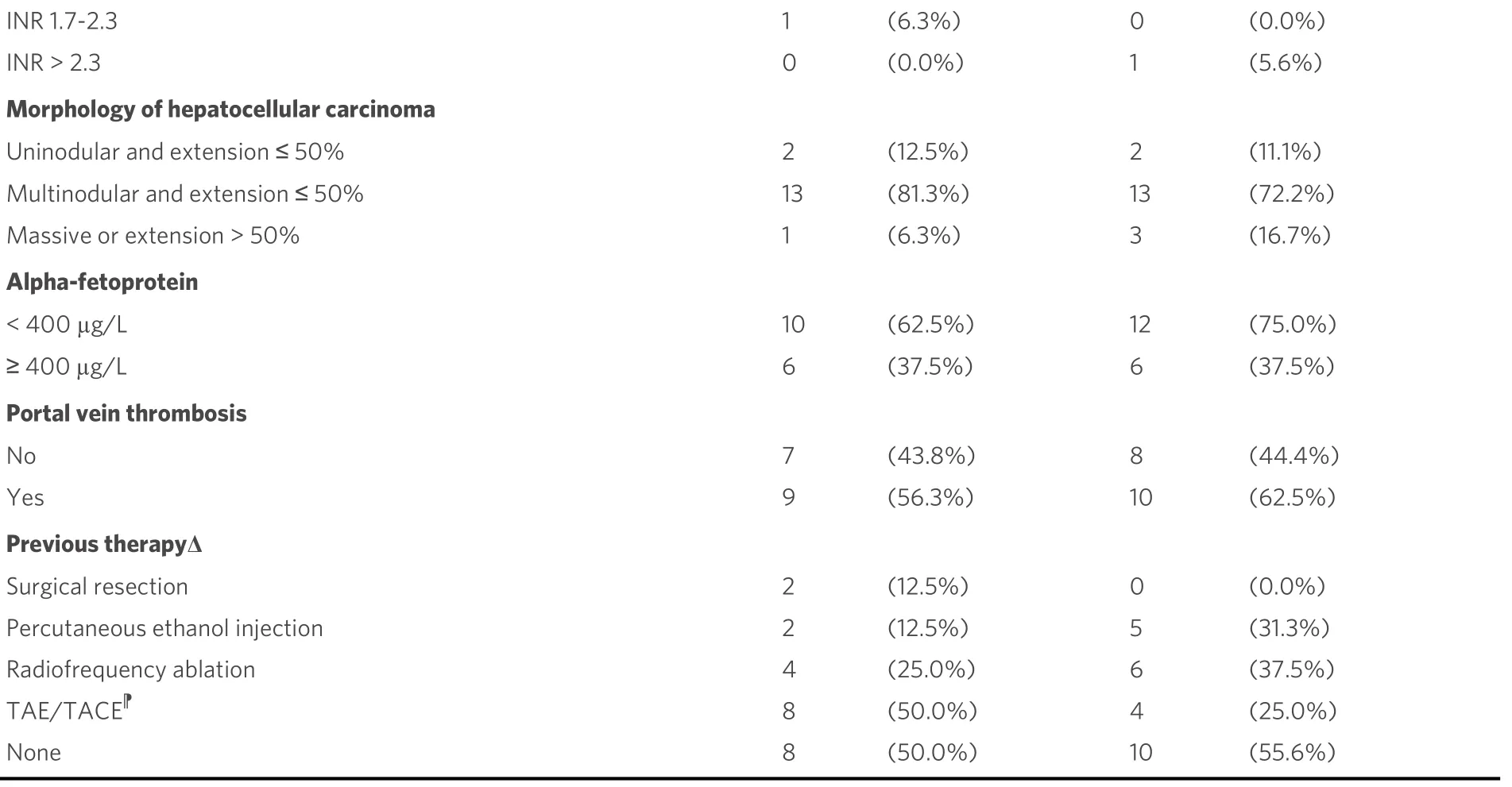
†BSC: Best supportive care; ‡CLIP: cancer liver Italian program; §ECOG: Eastern Cooperative Oncology Group; |: the same patient may have more than one aetiologic factors; Δ: the same patient may have received more than one previous treatment; TAE/TACE : transarterial embolization/transarterial chemoembolization.
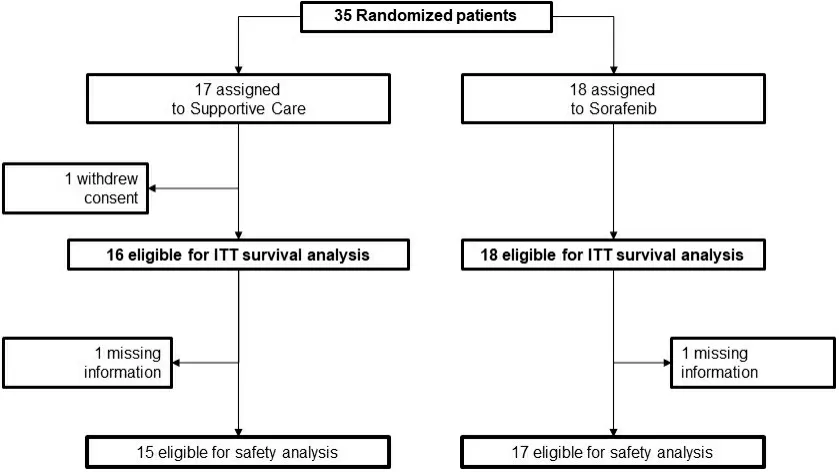
Figure 2. Study flow.
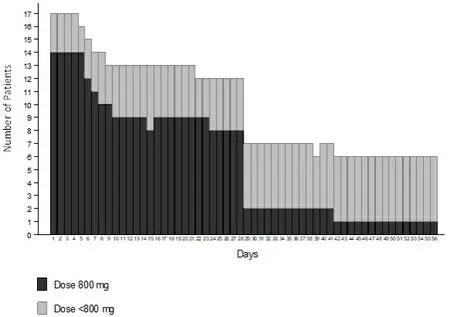
Figure 3. Profile of sorafenib dosing over the first two cycles.
Efficacy
With the database locked on 18 January 2019, 28 deaths were recorded, 12 in the control arm and 16 in the experimental arm: median OS was 4.9 (95%CI: 1.2-5.6) and 3.5 (95%CI: 1.3-5.3) months in the two arms,respectively [Figure 4A]. In the experimental arm, 6 patients progressed and 13 died without clinical or radiologic progression; median PFS was 2.4 (95%CI: 1.2-3.0) months [Figure 4B].
Safety
Safety analysis included 15 patients in the control and 17 in the experimental arm. There were no toxic deaths. At least one severe adverse event was reported in 2/15 (13.3%) patients in the control arm (always Grade 3), and in 9/17 (52.9%) in the sorafenib arm (always Grade 3, but with one case of Grade 4 AST increase). In addition, at least one adverse event (any grade) was reported in 6/15 (40.0%) patients in the control arm and in 15/17 (88.2%) in the sorafenib. Table 2 summarizes side-effects by grade and treatment arm.
QoL
Baseline QoL questionnaires were completed by 9 of 16 patients (56.2%) in the control arm and 12 of 18 patients (67%) in the experimental one. The mean global QoL score (Items 29 and 30 of the EORTC-C30)was 53.7 in the control arm and 62.5 in the experimental one. Thereafter, compliance dramatically decreased (with 3 and 4 questionnaires filled in after one cycle, respectively) and prevented any descriptionof the impact of treatment on QoL.

Table 2. Any grade and severe toxicity according to CTCAE categories and subcategories, by treatment arm
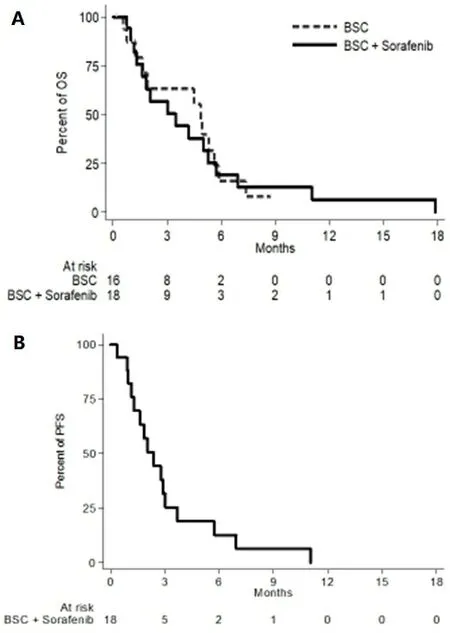
Figure 4. Overall survival (A) and progression-free survival (B) curves.
DISCUSSION
BOOST failed in its enrolment target. This was surprising for us, because the study was born within a clinical group that in the past had been extremely effective in conducting randomized clinical trials[16]. At the beginning of the study, the reason for the slow engagement of the investigators was surely the drug completely in charge of the local, enrolling institution. However, even later, when, thanks to the AIFA funding, the drug could be provided to local investigators free of charge, the enrollment did not proceed as expected. We believe that several reasons could have contributed to this. Above all, being sorafenib available in clinical practice since 2007, oncologists and hepatologists, thanks to clinical experience, might have reduced over time their uncertainty regarding the identification of candidate patients, excluding those with worse clinical conditions, or eventually intensifying supportive care to downstage Child-Pugh from B (score 7) to A, and attempting the treatment with sorafenib in patients with a strong motivation. In this context,several investigators could have lost their equipoise regarding the study question.
However, unfortunately, the study question remains unanswered.
The two registered trials, Sharp and Asia Pacific, enrolled only a few dozen Child-Pugh B patients (overall less than the BOOST study); therefore, in several countries, reimbursement of sorafenib is allowed only for Child-Pugh A patients[10].
Retrospective, uncontrolled, and real life studies on the efficacy of sorafenib in HCC have been reported[17-22]. The prospective observational phase 4 GIDEON trial was the largest one. It enrolled 3371 patients treated with sorafenib, including 666 Child-Pugh B and 74 Child-Pugh C cases. As expected,survival was longer for Child-Pugh A patients compared to Child-Pugh B ones (13.6 monthsvs. 5.2 months,respectively). Occurrence of serious adverse events (60%vs. 36%) and the rate of subsequent treatment discontinuation (40%vs. 29%) were higher in Child-Pugh B than in Child-Pugh A patients[21].
A prospective, observational, multicenter study (INSIGHT) enrolled 791 patients treated with sorafenib in daily practice conditions. In this trial, 182 (23%) Child-Pugh B patients were included. The median OS was 17.6, 8.1, and 5.6 months for Child-Pugh A, B, and C patients, respectively. In this study, adverse events were less frequent among Child-Pugh B and C patients (50.8% and 30.8%, respectively) compared to Child-Pugh A ones (72.4%), possibly due to the shorter duration of treatment[22].
In an Italian multicenter, open-label, phase 2 trial including Child-Pugh A (234 cases) and B patients (63 cases), median survival was shorter (3.8 monthsvs. 10.0 months) and the rate of adverse events was similar or lower for Child-Pugh B compared to Child-Pugh A patients[20].
Overall, compliance and toxicity data reported in the small group of patients treated within the BOOST study are consistent with other findings reported in the literature in the same setting.
Finally, McNamaraet al.[23]published a metanalysis of studies including HCC patients (both Child-Pugh A and B) treated with sorafenib. The results regarding 1684 Child-Pugh B patients are similar to those of the above-mentioned studies, with a worse prognosis than Child-Pugh A patients, due to their poorer liver function, but no conclusions on the efficacy of sorafenib might be drawn in these patients.
Despite the limitations of the present study, due to failure in reaching the enrolment goal, BOOST represents one of the few randomized lines of evidence available on the use of first-line sorafenib in Child-Pugh B HCC patients. Very recently, the results of the PRODIGE 21 phase 2 trial were published, suggesting a potential benefit of sorafenib in patients with HCC and Child-Pugh B liver cirrhosis with ALBI score 1/2[24]; however, this needs to be confirmed prospectively. Similar results have been shown with lenvatinibn[25], cabozantinib[26], and ramucirumab[27]. Overall, these data suggest that a good baseline liver function - as indicated by a lower ALBI score - is associated with a better outcome and a milder toxicity in patients with advanced HCC treated with the above drugs.
We could not calculate ALBI scores for our patients since albumin and bilirubin values were collected as categorical variables. Similar to what happened to our trial, Labeuret al.[28]reported the premature termination of a study aimed at personalizing sorafenib therapy in patients with HCC and Child-Pugh B7-8 liver function, assessing also sorafenib pharmacokinetics. However, only 5 out of the 45 planned patients were actually recruited, over a period of almost three years.Notwithstanding the very low statistical power of BOOST that does not allow drawing any firm conclusions,the lack of any positive efficacy signal and the very poor compliance to sorafenib, together with all the available evidence mentioned above, led us to believe that there is no more room for programming further similar trials. Unfortunately, the lack of generalizability of study results to Child-Pugh B patients will also affect new drugs in HCC. Patients with CP-B were either completely excluded from registered trials of regorafenib[29]and lenvatinib[30]or allowed only if the Child score was 7 in the nivolumab CheckMate 040 study (with only 49 patients actually enrolled in the expansion cohort)[31]. The analysis of this latter group of patients indicated that, similar to sorafenib, nivolumab can be administered to some patients with Child-Pugh B liver function. However, the efficacy of nivolumab in this setting remains to be demonstrated. A retrospective analysis of a subgroup of 73 patients randomized in the CELESTIAL trial (51 to cabozantinib and 22 to placebo) who progressed to Child-Pugh B class at Week 8 of treatment showed that, while the OS benefit of cabozantinib over placebo was maintained, some Grade 3/4 adverse events (fatigue, ascites, AST elevation, and thrombocytopenia) were more common in the cabozantinib arm, while others (palmarplantar erythrodysesthesia and hypertension) were more frequent in the placebo arm[32].
Thus, systemic treatment of advanced HCC patients with Child-Pugh B liver cirrhosis will remain an unmet need in the next years as well.
In conclusion, the BOOST trial failed and suggests that, when a wide registration of drugs (i.e., not fully supported by scientific evidence) has occurred, it may preclude advancing the knowledge of the actual efficacy of these drugs in selected populations. In fact, due to the availability of a drug in the general practice setting, neither will the investigators propose nor will the patients accept to be randomized to BSC. This scenario, as well as the BOOST results, should inspire researchers and the industry to plan clinical trials in definitive populations early in the development plan and the regulators to ask for more definitive evidence before clearing drugs for the general use in practice[33].
DECLARATIONS
AcknowIedgments
We thank Giuliana Canzanella, Marilena Martino, Maria Teresa Ribecco, Fiorella Romano, Giovanni de Matteis, Alfonso Savio, Lucia Sparavigna, Antonia Del Giudice, Simona Bevilacqua, Manuela Florio,Valentina Barbato, Anna Gimigliano for trial management at the Clinical Trial Unit of the Istituto Nazionale Tumori, IRCCS, Fondazione G.Pascale, Napoli.
Authors’ contributions
Contributed conception, design and implementation of the research, to the analysis and interpretation of the data and to the writing of the manuscript: Daniele G, Schettino C, Piccirillo MC, Gargiulo P, Perrone F,Gallo C, Arenare L, Daniele B
Contributed to acquisition and interpretation of data: Farinati F, Federico P, Tamberi S, Crivellari G,Barni S, Tortora R, Izzo F, Frassoldati A, Cavanna L, Mucciarini C, Bolondi L, Dinota A, Pelizzaro F,Di Maio M, Bilancia D
All the Authors give final approval of the manuscript to be submitted.
AvaiIabiIity of data and materiaIs
Data of this study will be shared with publication upon reasonable and motivated request to the Principal Investigator of the study (bruno.daniele@aslnapoli1centro.it). The following IPD will be available for sharing: baseline characteristics of patients, treatment data, safety data, follow-up data. There will be no time limit for data sharing.
FinanciaI support and sponsorship
The study was supported by the Italian Drug Agency (AIFA) in May 2011 (code FARM84SA2X) for trial coordination activities.
ConfIict of interest
Massimo Di Maio reports personal honoraria (for participation in meetings, advisory boards or for acting as consultant) from Bristol Myers Squibb, Merck Sharp & Dohme, Pfizer, AstraZeneca, Eisai, Takeda, Janssen,Astellas, and institutional research grant from Tesaro.
Antonio Frassoldati reports personal honoraria for advisory board from Novartis and Roche, invited lectures Novartis, Pfizer, Lilly, AstraZeneca, Celgene.
Sandro Barni reports personal honoraria from Genomic Health, Bayer AG, Eli-Lilly, Roche, Italfarmaco,Eisai, Astellas, Mylan, Leo Pharma, Pharmanutra, Medac Pharma, Kyowa Kirin.
Luigi Cavanna reports travel/Accomodations/meeting expenses from Pfizer, Ipsen, Celgene and personal honoraria for consultancy from AstraZeneca, Merck.
Raffaella Tortora reports personal honoraria for advisory boards from Bayer and Abbvie.
Gennaro Daniele: reports personal honoraria for advisory board from Beigene, and travel support from Roche.
Luigi Bolondi reports personal honoraria from Bayer, BMS, Sirtex, Guerbet, Alfa Sigma.
Francesco Perrone reports grants, personal fee and non-financial support from Bayer, grants and personal fees from Incyte and Astra Zeneca, personal fees from Celgene, Sandoz, Pierre Fabre, Janssen Cilag, Roche,and grants from Pfizer.
Maria Carmela Piccirillo reports personal fees from Daichii Sankyo, GSK, MSD, grants from Roche, grants and personal fees from AstraZeneca, non-financial support from Bayer.
Bruno Daniele reports personal fees from Bayer, Eli Lilly, Ipsen, Astrazeneca, MSD; Sanofi Aventis, Roche,personal fees and payment for lectures from Eisai.
The other authors declared that there are no conflicts of interest.
EthicaI approvaI and consent to participate
The study was promoted by the Istituto Nazionale per lo Studio e la Cura dei Tumori, IRCCS, Fondazione G. Pascale, Napoli, Italy. The study was performed in accordance with the Declaration of Helsinki. Ethics Committees at each participating Institution approved the study, and all the patients signed the informed consent before any study related procedure.
Consent for pubIication
Not applicable.
Copyright
© The Author(s) 2021.

- Hepatoma Research的其它文章
- A practical approach to pediatric liver transplantation in hepatoblastoma and hepatocellular carcinoma
- 3D Organoid modelling of hepatoblast-like and mesenchymal-like hepatocellular carcinoma cell lines
- Effect of mesenchymal stem cell in liver regeneration and clinical applications
- Prognostic factors associated with survival in patients with hepatocellular carcinoma undergoing transarterial chemoembolisation: an Australian multicenter cohort study
- AUTHOR INSTRUCTIONS