
利用荧光SSR分子标记评估中国栗属植物遗传多样性
2021-05-07 06:28:20聂兴华郑瑞杰赵永廉曹庆芹秦岭邢宇
中国农业科学 2021年8期
聂兴华,郑瑞杰,赵永廉,曹庆芹,4,秦岭,4,邢宇,4
利用荧光SSR分子标记评估中国栗属植物遗传多样性

1北京农学院植物科学技术学院,北京 102206;2辽宁省经济林研究所,辽宁大连 116000;3北京市怀柔区板栗技术试验与推广站,北京 102206;4林木分子设计育种高精尖创新中心,北京 102206
【】利用SSR分子标记研究中国栗属植物遗传多样性、亲缘关系和群体遗传结构的特点,为栗属植物的资源改良、种质创新与利用提供理论依据。利用不同产区的12个板栗品种对330个SSR分子标记进行筛选,获得高质量的12对SSR引物。随后在高分辨率的毛细管电泳上对栗属4个种的96份资源进行位点信息检测。用Power Marker 3.25、GenAlEx 6.51、FigTree v1.4.3和Structure 2.3.3对全部资源进行群体遗传多样性的相关分析。对96份资源进行检测,共获得129个等位变异,每个标记平均有10.750个位点变异。位点多样性(GD)变幅为0.656(CmSI0396)—0.877(CmSI0930),平均为0.800;观察杂合度(Ho)变幅为0.329(CmSI0742)—0.769(CmSI0702),平均为0.615;期望杂合度(He)变幅为0.489(CmSI0742)—0.789(CmSI0922),平均为0.672;多态信息含量(PIC)变幅为0.586(CmSI0396)—0.868(CmSI0930),平均为0.774。从不同栗属植物种群间的遗传多样性来看,茅栗种群的观察位点数(Na)、有效等位变异(Ne)和Shannon多样性指数最高,其次是板栗种群,最低是日本栗种群。从两两群体间的遗传分化指数(Fst)可知,栗属植物种间的遗传分化值在0.077—0.180,整体种群间存在中等以上程度的分化,板栗种群、锥栗种群与日本栗种群间的遗传分化值分别为0.165和0.180,表现出较大的遗传分化。同时,栗属植物种群的基因流(Nm)为1.580>1,也说明种群间存在较频繁的基因交流,由此降低了由基因遗传漂变所引起的各种群间遗传分化程度。分子方差分析(AMOVA)结果表明,变异主要发生在种群内,占总变异量的73%,种群间的变异占27%。UPGMA聚类分析、主坐标分析和群体遗传结构结果较一致,各资源的遗传背景存在明显的种间界限,部分资源在世代遗传中继承了不同祖先种的遗传信息。例如,资源65、71和82号为混合类型资源,包含有茅栗和日本栗的遗传背景,而现在的两种间存在地理隔离。在相同生态区域的栗属植物种间存在一定的基因交流,没有形成完全的生殖隔离。48号资源‘广东矮生’同时含有板栗和茅栗的遗传背景,在地理分布上该资源原生地正处于板栗和茅栗资源的重叠生态区。筛选的12对SSR引物能够准确地评估中国栗属植物的遗传多样性,综合聚类分析可确定栗属植物的类群划分与种间信息高度一致且种间存在一定的基因交换。
SSR;栗属;聚类分析;群体遗传结构;群体遗传分化
0 引言
【研究意义】目前世界公认的栗属植物有板栗(Bl.)、锥栗((Skam) Rehd. et Wils.)、茅栗(Dode)、日本栗(S. et Z.)、欧洲栗(Mill.)、美洲栗((Marsh.) Brokh.)和美洲榛果栗(Mill.)7个种[1]。20世纪以来,墨水病和栗疫病严重打击了曾经盛极一时的欧洲栗和以材用为主的美洲栗,造成欧洲栗出现生产危机和几乎全境的美洲栗濒于灭绝[2]。中国分布板栗、茅栗、锥栗和日本栗(辽东栗)4个种的资源,且分布十分广泛,跨越温带、暖温带和亚热带,同时我国栗属资源对栗疫病、墨水病等表现出较好的抗性,具有最为丰富的遗传多样性[2-3]。由于长期的自然和人工选择,中国栗属植物不仅具有很高的食用品质,还为世界各地栗属植物的基因改良提供了重要的来源,在世界栗属种质资源中占有不可代替的地位[4]。因此,了解中国栗属种质资源的亲缘关系,评估各种间资源的遗传多样性和群体遗传结构,对栗属植物优异种质的创新与开发有重要的意义。【前人研究进展】目前,研究者们已经开发出了诸多第二代和第三代分子标记,并将其广泛应用于植物的遗传多样性、遗传图谱和基因组分析中[5-7]。例如随机扩增的多态性DNA(RAPD)、限制性片段长度多态性(RFLP)、简单序列重复(SSR)和三代标记单核苷酸多态性(SNP)等。其中,SSR串联重复序列的突变率非常高,能形成高度多态的等位变异[8]。此外,SSR标记具有共显性、稳定性和可重复性,可从物种的基因组和转录组测序中开发获得。在栗属植物的最新研究中,SSR主要用于遗传多样性研究、关联分析和遗传图谱的构建等领域。INOUE等[9]利用17个SSR标记成功进行了中国板栗、日本栗和欧洲栗跨种间的扩增,3个物种中的65个样品表现出相当高的遗传多样性。JIANG等[10]对中国10个省的95个板栗品种进行基于SSR的分析,评估了中国板栗的遗传多样性,遗传结构模式和连锁不平衡(LD)。MULLER等[11]利用24个SSR标记对272个美洲栗个体进行基因分型,发现了5个特异的位点与种群所在地的环境变量存在显著的相关性。KUBISIAK等[12]创建了一个基于转录组测序开发的330个SSR和1 071个SNP标记组成的板栗遗传图谱,通过QTL关联分析筛选获得3个抗病区段。此外,刘国彬等[13]利用13对SSR分子标记对北京怀柔区的33株明清古板栗树进行了指纹图谱构建。【本研究切入点】中国作为栗属植物的遗传多样性中心,丰富的栗属植物资源在农业生态建设和区域农业经济创收方面发挥着重要作用。但栗属植物优异种质的开发与利用还有很大的局限性,以往的研究多集中在单个种的遗传多样性,而对中国栗属植物的遗传多样性研究相对较少。【拟解决的关键问题】本研究利用高质量的SSR标记评估96份中国栗属植物资源的遗传多样性,并解析不同栗属种质资源的亲缘关系、群体遗传结构和遗传背景,为栗属植物优异种质的创新与开发提供理论依据。
1 材料与方法
1.1 筛选引物的材料
试验中筛选引物所需的12份板栗品种材料于2019年在北京市怀柔区板栗技术试验与推广站进行选取(电子附表1)。
1.2 试验材料
试验中供试材料于2019年在我国不同地区的资源圃和野外进行收集,共计96份(表1、电子附表2),包括板栗、锥栗、茅栗和日本栗4个种。其中板栗50份,采自北京市怀柔区板栗技术试验与推广站;锥栗13份,采自福建省建瓯市锥栗接穗圃;野生茅栗资源19份,采自安徽、江西、湖南和湖北等省的野外;日本栗14份,采自辽宁省经济林研究所栗属植物资源圃。每份材料采集嫩叶6片,液氮处理后,保存于-80℃超低温冰箱备用。
1.3 方法
1.3.1 DNA提取 板栗基因组DNA提取方法采用本实验室的CTAB改良方法[14]。提取DNA后,使用1%琼脂糖凝胶电泳和Nano Drop One分光光度计(Thermo Fisher Scientific,USA)分别测试DNA的质量和浓度。然后将每份DNA的浓度稀释至20—50 ng∙μL-1以备PCR扩增使用。

表1 供试材料的地理分布
1.3.2 EST-SSR分子标记的来源 SSR分子标记来源于美国林木基因组(https://www.hardwoodgenomics. org/)公布的中国板栗EST-SSR库。利用聚丙烯酰胺凝胶电泳检测技术对数据库中的330个EST-SSR标记进行初筛,引物由擎科生物科技有限公司合成。
1.3.3 普通SSR-PCR扩增体系 PCR扩增采用10 μL反应体系:4 μL ddH2O,4 μL TaKaRa公司生产的2×Taq PCR Master Mixes,上、下游引物各0.5 μL,1 μL模板DNA(20—50 ng∙μL-1)。反应程序为:94℃预变性3 min;94℃变性30 s,56℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min,10℃保存。
1.3.4 PAGE凝胶制备与位点信息检测 凝胶检测参照本试验室优化的PAGE凝胶制备与位点检测方法[15]。
1.3.5 荧光毛细管电泳SSR-PCR扩增体系 PCR扩增采用20 μL反应体系:其中包含1 μL模板DNA(20—50 ng∙μL-1),10 μL 2×Taq PCR Master Mixes(Takara,DaLian China),0.1 μL正向引物(10 μmol∙L-1),0.3 μL反向引物(10 μmol∙L-1),0.2 μL荧光标记(FAM,HEX,ROX或TAMRA)的M13引物(10 μmol∙L-1)(中国天津,擎科)和9.4 μL ddH2O。反应程序为:94℃预变性3 min;94℃变性30 s,56℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min,10℃保存(BIO-RAD PCR Thermal Cycler T100,USA)。
1.3.6 荧光毛细管电泳位点信息检测 将3对或4对荧光SSR引物的PCR扩增产物合并在一起。然后使用ABI 3730XL DNA测序仪(Applied Biosystems,Foster City,CA,USA)通过毛细管电泳分析混合物中片段位置信息,并使用Gene Marker v 2.2.0软件(Soft Genetics LLC,State College,PA,USA)读取等位变异具体的片段大小信息。
1.4 数据分析
使用Power Marker 3.25[16]和GenAlEx 6.51[17-18]软件计算包括主要等位基因频率(MAF)、等位基因数量(Na)、基因多样性(GD)、杂合性(He)、Shannon多样性指数(I)、多态信息含量(PIC)等遗传多样性指标。使用Power Marker 3.25计算各资源间的Nei’s遗传距离,随后基于UPGMA的方法在FigTree v1.4.3中构建聚类树。使用Structure 2.3.3[19]进行群体结构的分析。通过GenAlEx 6.51计算种群间的遗传分化固定指数(Fst)、基因流(Nm)、主坐标分析(PCoA)和分子变异分析(AMOVA),利用这些数据评估各种群间的遗传分化和遗传变异特征。
2 结果
2.1 高质量SSR引物的筛选
本试验选取不同产区的12个板栗品种作为筛选阶段的材料,以分布于不同板栗连锁群的330对SSR引物为基数,通过聚丙烯酰胺凝胶电泳筛选高质量和高多态的SSR引物作为后续进行遗传多样性分析的备选引物。以条带清晰、位点多态性高为原则,最终在330对引物中筛选出12对高多态引物。12对SSR引物分别是CmSI0396、CmSI0561、CmSI0614、CmSI0658、CmSI0702、CmSI0742、CmSI0800、CmSI0853、CmSI0871、CmSI0883、CmSI0922、CmSI0930(表2),图1是引物CmSI0561和CmSI0614的位点信息图例,通过分型统计可知CmSI0561在12个板栗品种中有6个不同的位点组合,CmSI0614有7个不同的位点组合,表现出高多态性。这些多态的SSR引物进一步用高分辨率的毛细管电泳进行位点检测。
2.2 SSR分子标记的遗传多样性
选取12对高质量SSR标记,采用荧光毛细管电泳技术对96份栗属植物资源进行分析(表3)。相比凝胶电泳检测,毛细管电泳检测的分辨率能达到1 bp,更能准确地读取位点信息(图2),为了检验毛细管电泳检测的准确性,本试验先用聚丙烯酰胺凝胶电泳与毛细管电泳进行检测对比。以引物CmSI0614为例(图2),毛细管电泳检测结果与凝胶电泳检测位点一致,并且能准确地读取具体的片段大小。

图1 高多态引物CmSI0561和CmSI0614在12个板栗品种中的位点信息

表2 12对高质量SSR标记的信息表
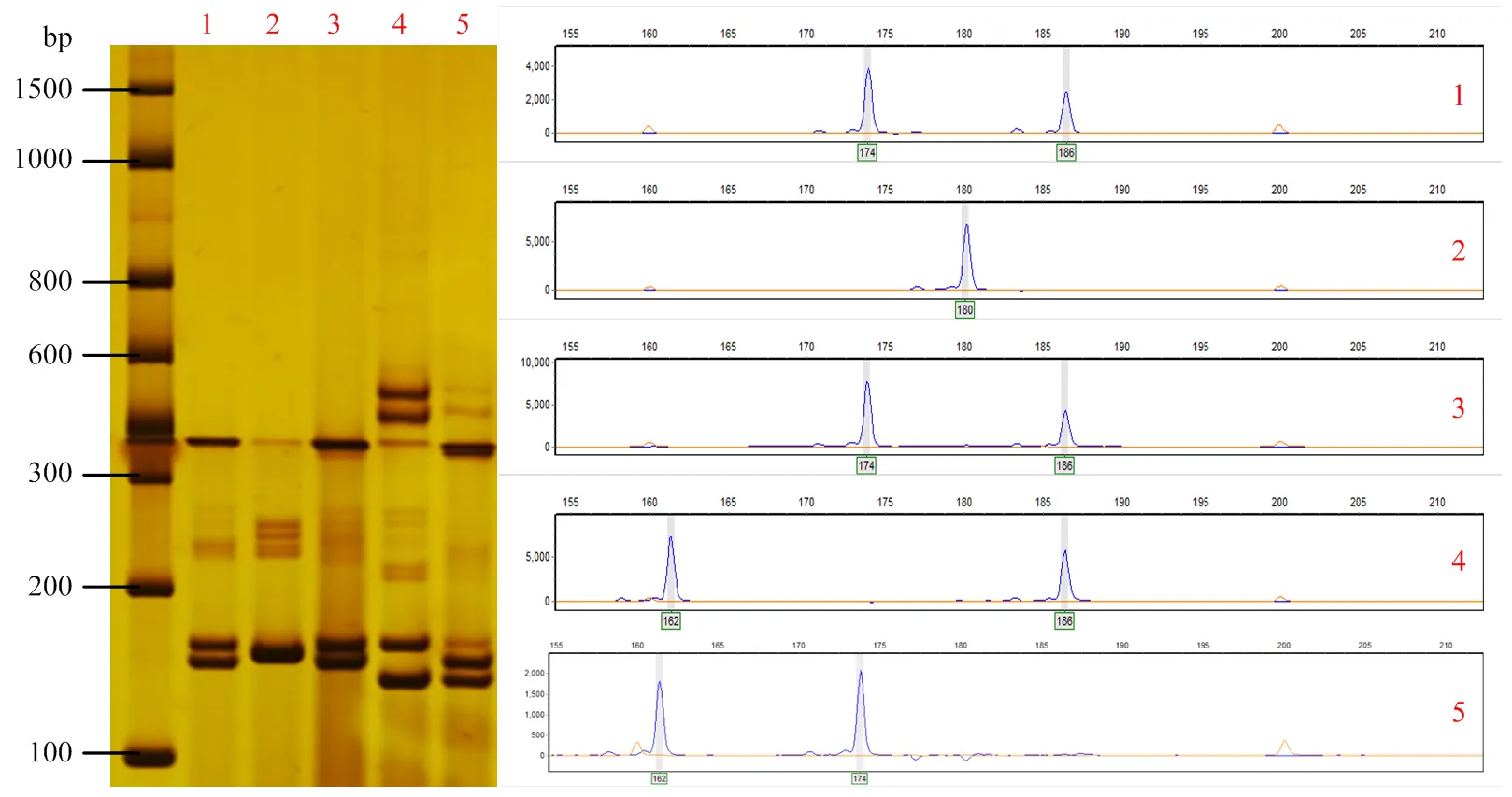
图2 聚丙烯酰胺凝胶电泳银染结果和毛细管电泳检测结果对比
利用毛细管电泳检测共获得129个等位变异,每个标记平均10.750位点变异。等位变异的大小范围从标记CmSI0742的137 bp到标记CmSI0922的351 bp。位点多样性(GD)变幅为0.656(CmSI0396)—0.877(CmSI0930),平均为0.800;观察杂合度(Ho)变幅为0.329(CmSI0742)—0.769(CmSI0702),平均为0.615;期望杂合度(He)变幅为0.489(CmSI0742)—0.789(CmSI0922),平均为0.672;多态信息含量(PIC)变幅为0.586(CmSI0396)—0.868(CmSI0930),平均为0.774;单个分子标记层面的Fst指数变幅为0.066(CmSI0853)—0.304(CmSI0742),平均为0.172。
2.3 栗属植物种群间的遗传多样性
分析不同栗属植物种群间的遗传多样性数据(表4),可知茅栗种群的观察位点数(Na)、有效等位变异(Ne)和Shannon多样性指数均为最高,其次是板栗种群,最低的是日本栗种群;在杂合度方面,茅栗也表现出较高的杂合性。茅栗种群有最为丰富的遗传多样性,这可能与其资源类型有着密切关系,茅栗是未开发的野生种质资源,而选用的板栗、锥栗和日本栗为实生选育的品种或农家种质资源。
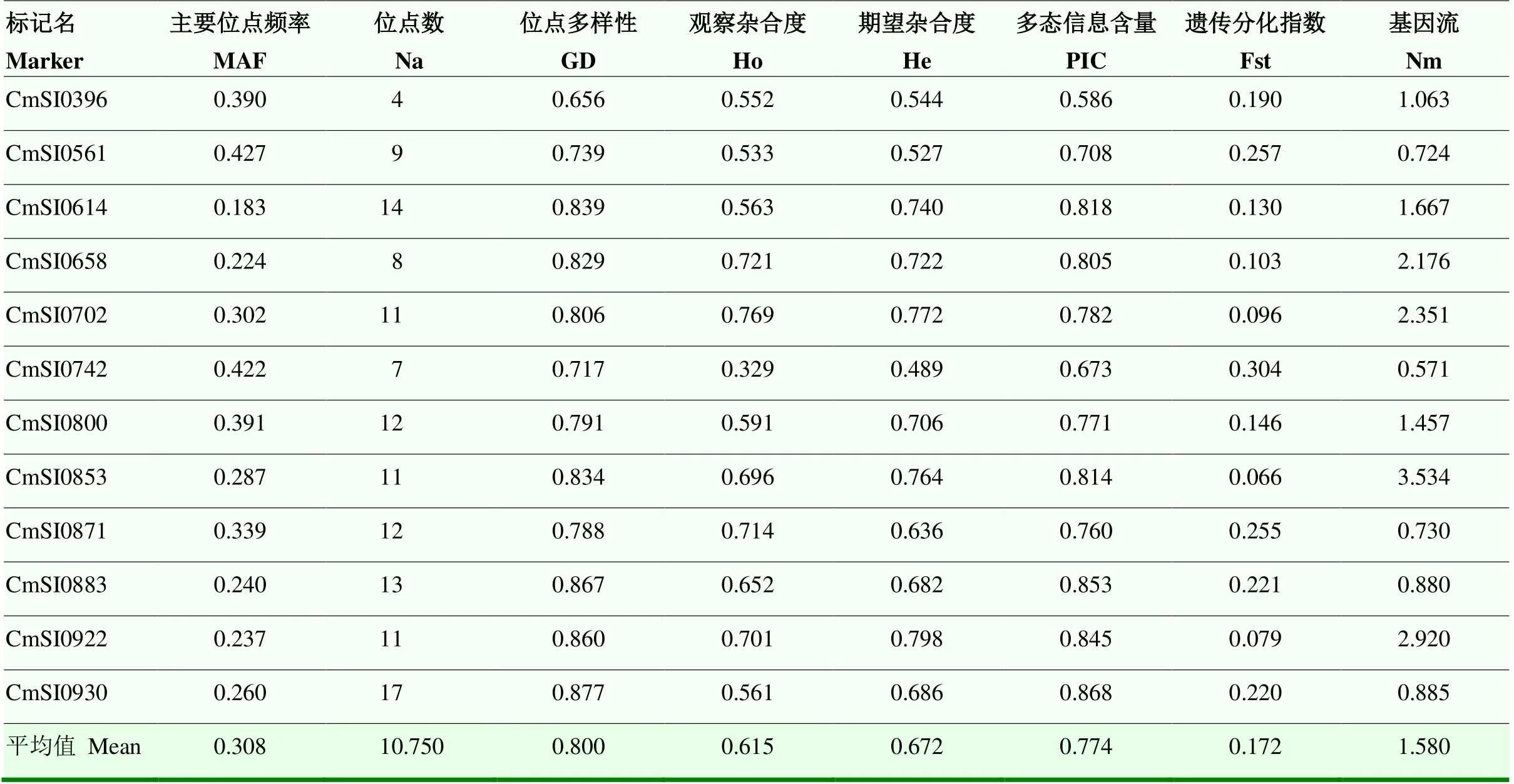
表3 12对SSR标记的关键遗传数据

表4 不同栗属植物种群间的关键遗传数据
2.4 栗属种群遗传分化与变异分布
通过计算两两群体间的Fst固定系数(表5),可知板栗种群与锥栗种群、茅栗种群间的遗传分化值在0.05—0.15,种群间存在中等程度的分化,而与日本栗种群间的遗传分化值为0.165,表现出较大的遗传分化。锥栗种群与茅栗种群间的遗传分化值为0.115,群体间存在中等程度的分化,与日本栗种群间的遗传分化值为0.180,也表现出较大的遗传分化。茅栗种群与日本栗种群间的遗传分化值0.108,种群间存在中等程度的分化。该结果与单个分子标记层面Fst值的结论较为一致。
为了确定栗属植物种群间位点的变异总量及在种群间和种群内的变异分布,本试验对4个种群进行了分子方差分析(AMOVA)。结果表明,变异主要发生在种群内,占总变异量的73%,种群间的变异占27%。同时,栗属植物种群的基因流(Nm)为1.580,大于1,也说明种群间存在着一定的基因交流,由此降低了因遗传漂变引起的各种群间遗传分化程度。

表5 两两种群间的Fst值
2.5 栗属植物的群体结构和主坐标分析
通过对96份资源进行群体遗传结构分析(图3),结果表明,当K=3时,Delta K出现明显峰值,将供试材料划分为3个组群(图4)。其中绿色组群(50份)主要为板栗种群,代表板栗的主要基因库;蓝色组群(13份),全部为锥栗种群,代表锥栗的主要基因库;红色组群(33份),主要为茅栗和日本栗资源,同时部分资源为种间混合类型。当K=4时,也表现较高的Delta K值,此情况下供试材料中的日本栗资源独立出来(图4),由此可判断日本栗和茅栗种间具有较近的亲缘关系。
通过对供试材料进行主坐标分析(PCoA),可知全部资源可大致划分为4个组,主坐标(PCo)1和2分别解释了位点信息数据中20.89%和8.76%的变异(图5)。红色代表板栗资源,大部分资源位于左上部成为一簇;绿色代表锥栗资源,都位于中下偏左部成为一簇;蓝色代表茅栗资源,大部分资源位于中下偏右部成为一簇;紫色代表日本栗资源,都位于右上部成为一簇。同时,在PCoA可以观察到,个别锥栗资源与板栗资源,个别茅栗资源与日本栗资源相互掺杂,结合群体结构分析可知这些资源为种间混合类型。

图3 供试栗属植物群体结构分析的Delta K值分布
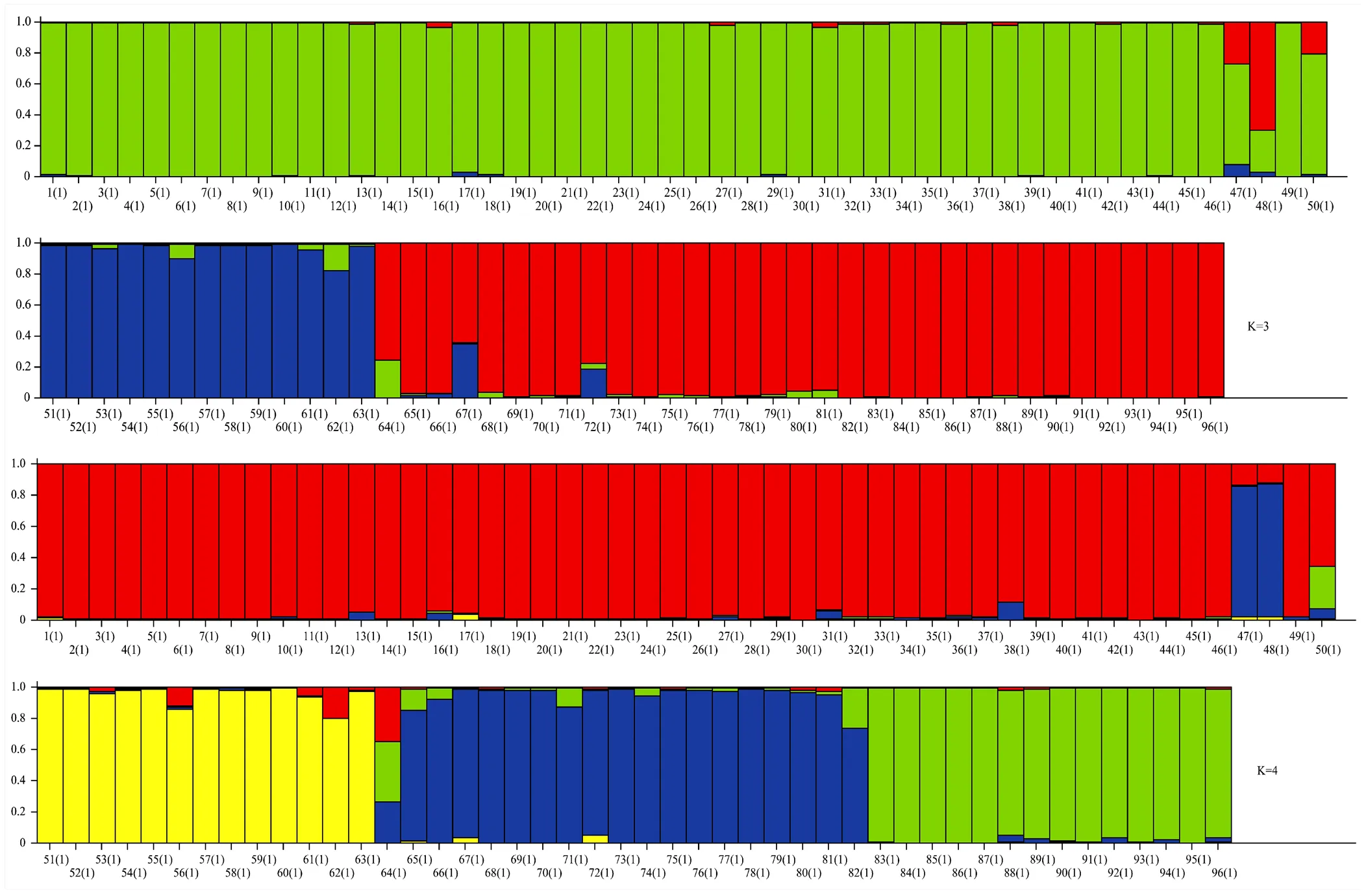
图4 供试栗属植物的群体结构图

表6 栗属植物种群间的AMOVA分析

图5 供试栗属植物的主坐标分析(PCoA)
2.6 不同栗属资源的聚类
通过对96份资源进行基于UPGMA方法的聚类分析(图6),结果表明,全部资源被划分为4个类群(Pop1、2、3、4),全部的50份板栗资源聚类在Pop1中;Pop2共计13份,全部是锥栗资源;Pop3共计16份资源,主要是茅栗资源;Pop4共计17份资源,包括全部日本栗资源和3个茅栗资源。Pop4中3个茅栗资源与日本栗资源独立分开成立一支,结合群体结构的结果,可知3个茅栗资源中有日本栗基因的混入。供试材料的聚类结果与群体结构分析和主坐标分析结果基本一致,进一步说明茅栗与日本栗之间具有较近的亲缘关系。
3 讨论
3.1 SSR分子标记的筛选及其在栗属植物遗传多样性方面的运用
SSR分子标记具有稳定性、共显性、可重复性和多态性的特点,被广泛应用于各种遗传相关领域,例如资源鉴定、遗传图谱、遗传多样性以及核心种质的选择等[20-21],这些研究的有效性和成功与否很大程度上取决于选用的分子标记的质量和基因型数据的准确性[22-23]。MULLER等[11]报道了利用24个SSR标记对272个美洲栗个体进行基因分型,共获得135个等位变异,平均每对引物5.6个。江锡兵等[10]利用17个SSR分子标记在来自10省份的95个板栗品种共获得44个位点变异,平均每对引物2.6个,多态性信息含量(PIC值)平均为0.352。张馨芳等[24]利用21个SSR标记对151份板栗资源进行检测,共获得71个变异位点,平均每对引物3.38个,多态性信息含量(PIC值)平均约为0.867,有效等位基因数(Ne)和Shannon多样性指数(I)分别为1.498、0.4655。黄武刚等[25]用10对SSR引物对69个野生板栗个体进行分析,共检测到84个等位变异,每位点的平均等位变异为8.400个,平均有效等位基因数(Ne)、平均期望杂合度(He)、平均多态性信息含量(PIC值)分别为4.998、0.777和0.739。本研究从330个SSR标记中筛选出12对高质量的引物,在收集的96个栗属植物中共获得129个等位变异,每对引物平均有10.750个位点变异,这是JIANG等[10]研究结果的近5倍,也高于张馨芳等[24]和黄武刚等[25]的研究结果。本研究使用12个SSR标记计算全部资源的观察杂合度(Ho平均值为0.615,显示出较高的杂合度,与前人研究结果一致[26],板栗的高度杂合特性与其为异花授粉植物密不可分[27-29]。12个SSR标记多态信息含量(PIC)的平均值为0.774(>0.500),最大值达到0.868,代表供试材料具有高多态性。其原因可能是本试验收集了更为广泛的种间种质资源,此外,本试验利用毛细管电泳检测技术进行读取和记录目的条带,不仅极大地提高了基因座检测的准确性,而且大大克服了凝胶电泳检测分辨率低的问题。

图6 供试栗属植物的UPGMA系统树谱图
3.2 中国栗属植物种间遗传多样性的差异
现今中国分布的栗属植物资源中板栗的栽培面积最大,共有5大品种生态群,其次是锥栗和日本栗,而具有童期短和矮化特性的茅栗处于待开发状态[2]。栗属植物作为典型的果树材料,由于其本身遗传特征的制约,栗属植物品种选育仍以实生选育为主,缺乏优质高效的品种和抗性良好的砧木种质资源。Shannon多样性指数(I)结果表明,茅栗种群最高,且茅栗的遗传多样性最高,这一结果与郎萍等[27]的结论不一致,其可能的原因有两个:其一,样品的选择不同,郎萍等选择的样品主要集中在长江中下游和西南地区,本研究中茅栗和板栗样品的选择覆盖度更广;其二,分子标记的技术不同,郎萍等利用的技术为同工酶,评估样品多样性的能力有限。本研究利用的技术是荧光SSR标记,其具有更丰富的多态性,能较好地反映样品的多样性。同时,茅栗是未开发的野生资源,茅栗资源可能保留着更为丰富的遗传变异,其巨大的种质利用潜力一直被人们所忽视。
在种间遗传分化方面,可知栗属植物种间的遗传分化值在0.077—0.180,种群间存在中等以上程度的分化,尤其板栗种群、锥栗种群与日本栗种群间表现出较大的遗传分化,这一结果与主坐标分析(PCoA)和群体遗传结构分析的结果具有高度一致性。同时,通过基因流(Nm)和分子方差分析(AMOVA)可知,栗属植物种群间基因流为1.580,表现出较高频繁的基因流动,但显著低于张馨芳等[24](Nm=9.696)及黄武刚等[25](Nm=2.476)在板栗种内利用SSR分子标记计算的基因流,也低于郎萍等[27](Nm=2.043)在板栗、锥栗和茅栗种间利用同工酶计算的基因流;栗属植物种群间的遗传变异占27%,也显著高于张馨芳等[24](4.9%)和黄武刚等[25](13%)在板栗种内居群间的遗传变异,而低于程丽莉等[30]通过cpSSR计算的栗属种间的遗传变异(33%)。因此,推测栗属植物作为一种典型的异花授粉植物,种群间在自然条件下具有一定的基因交换,正是这些基因流阻止了各种间的进一步分化,栗属的这种分化特点与WRIGHT[31]的理论相符。
在栗属植物种间群体遗传多样性研究方面,现阶段的研究主要集中在板栗或锥栗等单个种内不同地域的群体遗传多样性方面[31-34],而栗属植物种间群体遗传多样性的研究知之甚少。本研究通过群体遗传结构分析和UPGMA系统发育树分析,可知板栗与锥栗,锥栗与茅栗的遗传背景存在明显的种间界限,这与韩继承等[35]研究结果一致。同时板栗和锥栗、板栗和茅栗、茅栗和日本栗之间有着广泛的基因交流,在供试材料中有部分资源为种间混合类型。例如65、71和82号资源既有茅栗的遗传背景又有日本栗的遗传背景,在聚类树结构中位于茅栗和日本栗亚分支的中间位置。48号资源‘广东矮生’同时含有板栗和茅栗的遗传背景,在地理分布上,广东省正处于板栗和茅栗资源的重叠生态区;在园艺学性状上,其也表现出茅栗树势矮小和板栗坚果大的性状特征。综上可知,栗属植物种群间的遗传分化程度在中等以上,部分资源在世代遗传中继承了不同祖先种的遗传信息;在相同生态区域的栗属植物种间存在一定的基因交流,没有形成完全的生殖隔离。
4 结论
筛选的12对SSR引物能够准确评估中国栗属植物的遗传多样性,综合聚类分析可确定栗属植物的类群划分与种间信息高度一致且种间存在一定的基因交换,茅栗与日本栗具有较近的亲缘关系,茅栗遗传多样性较高,具有很大的利用潜力。这将为未来栗属植物种质创新提供重要的理论依据。
[1] 张宇和, 柳鎏, 梁维坚, 张育明. 中国果树志, 板栗, 榛子卷. 中国林业出版社, 2005: 22-27.
ZHANG Y H, LIU L, LIANG W J, ZHANG Y M. China Fruits’ Monograph, Chinese Chestnut: Hazelnut Volume. Beijing: China Forestry Publishing House, 2005: 22-27. (in Chinese)
[2] HUANG H W, DANE F , NORTON J D. Allozyme diversity in Chinese, Seguin and American chestnut (spp.). Theoretical and Applied Genetics, 1994, 88(8): 981-985.
[3] PEREIRALORENZO S, RAMOSCABRER A M. Chestnut, an Ancient Crop with Future. Production Practices and Quality Assessment of Food Crops Volume 1Springer Netherlands, 2004, 2: 105-161.
[4] LANG P, DANE F, KUBISIAK T L. Phylogeny of Castanea (Fagaceae) based on chloroplasttrnT-L-F sequence data. Tree Genetics & Genomes, 2006, 2(3): 132-139.
[5] 魏潇, 章秋平, 刘宁, 张玉萍, 徐铭, 刘硕, 张玉君, 马小雪, 刘威生. 不同来源中国李(L.)的多样性与近缘种关系. 中国农业科学, 2019, 52(3): 568-578.
WEI X, ZHANG Q P, LIU N, ZHANG Y P, XU M, LIU S, ZHANG Y J, MA X X, LIU W S. Genetic diversity of theL. from different sources and their related species. Scientia Agricultura Sinica, 2019, 52(3): 568-578. (in Chinese)
[6] 李剑锋, 张博, 全建章, 王永芳, 张小梅, 赵渊, 袁玺垒, 贾小平, 董志平. 基于SSR标记的谷子主要农艺性状关联位点检测及等位变异分析. 中国农业科学, 2019, 52(24): 4453-4469.
LI J F, ZHANG B, QUAN J Z, WANG Y F, ZHANG X M, ZHAO Y, YUAN X L, JIA X P, DONG Z P. Associated loci detection and elite allelic variations analysis of main agronomic traits in foxtail millet (L.) based on SSR markers. Scientia Agricultura Sinica, 2019, 52(24): 4453-4469. (in Chinese)
[7] 石甜甜, 何杰丽, 高志军, 陈凌, 王海岗, 乔治军, 王瑞云. 利用EST-SSR评估糜子资源遗传差异. 中国农业科学, 2019, 52(22): 4100-4109.
SHI T T, HE J L, GAO Z J, CHEN L, WANG H G, QIAO Z J, WANG R Y. Genetic diversity of common millet resources assessed with EST-SSR markers. Scientia Agricultura Sinica, 2019, 52(22): 4100-4109.(in Chinese)
[8] GUR-ARIE R, COHEN C J, EITAN Y, SHELEF L, HALLERMAN E M, KASHI Y. Simple sequence repeats in escherichia coli: Abundance, distribution, composition, and polymorphism. Genome Research, 2000, 10(1): 62-71.
[9] INOUE E, NIN L, HARA H, RUAN S A, ANZAI H. Development of simple sequence repeat markers in Chinese chestnut and their characterization in diverse chestnut cultivars.Journal of the American Society for Horticultural Science, 2009, 134(6): 610-617.
[10] JIANG X B, TANG D, GONG B C. Genetic diversity and association analysis of Chinese chestnut (Blume) cultivars based on SSR markers. Brazilian Journal of Botany, 2017, 40: 235-246.
[11] MULLER M, NELSON C.D, GAILING O. Analysis of environment- marker associations in American chestnut. Forests, 2018, 9(11): 695.
[12] KUBISIAK T L, NELSON C D, STATON M E, ZHEBENTYAYAYEVA T, SMITH C, OLUKOLU B A, FANG C, HEBARD F V, ANAGNOSTAKIS S, Wheeler N, SISCO P H, ABBOTT A G, SEDEROFF R R. A transcriptome-based genetic map of Chinese chestnut () and identification of regions of segmental homology with peach (). Tree Genetics & Genomes, 2013, 9(2): 557-571.
[13] 刘国彬, 曹均, 兰彦平, 王金宝. 明清板栗古树指纹图谱的构建. 江西农业大学学报, 2017, 39(1): 134-139.
LIU G B, CAO J, LAN Y P, WANG J B. Construction of SSR fingerprint on 33 ancient chestnut trees. Acta Agriculturae Universitatis Jiangxiensis, 2017, 39(1): 134-139. (in Chinese)
[14] 曹庆芹, 徐月, 冯永庆. 板栗基因组DNA不同提取方法的比较. 中国农学通报, 2007, 23(6): 160-163.
CAO Q Q, XU Y, FENG Y Q. Comparison studies on extracting genomic DNA of chestnut (BL) by different methods. Chinese Agricultural Science Bulletin, 2007, 23(6):160-163. (in Chinese)
[15] 聂兴华, 张卿, 刘阳, 代月星, 曹庆芹, 秦岭. 板栗SSR标记研究中聚丙烯酰胺凝胶银染显影体系的优化. 北方园艺, 2019(12): 27-33.
NIE X H, ZHANG Q, LIU Y, DAI Y X, CAO Q Q, QIN L. Optimization of PAGE silver-stained system based on SSR markers in the chestnut. Northern Horticulture, 2019(12): 27-33.(in Chinese)
[16] LIU K J, MUSE S V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics, 2005, 21(9): 2128-2129.
[17] PEAKALL R, SMOUSE P E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics, 2012, 28(19): 2537-2539.
[18] PEAKALL R, SMOUSE P E. GenALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 2006, 6: 288-295.
[19] FALUSH D, STEPHENS M, PRITCHARD J K. Inference of population structure using multi-locus genotype data: Linked loci and correlated allele frequencies.Genetics, 2003, 164: 1567-1587.
[20] TAUTZ D, RENZ M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Research, 1984, 12: 4127-4138.
[21] SARDAR S S, PRADHAN K, SHUKLA R P, SARASWAT R, SRIVASTAVA A, JENA S N, DAS A B. In silico mining of EST-SSRs inL. and their utilization for genetic structure and diversity analysis in cultivars/breeding lines in Odisha, India. Molecular Breeding, 2016, 36: 49.
[22] LIU S R, LIU H W, WU A L, HOU Y, AN Y L, WEI C L. Construction of fingerprinting for tea plant () accessions using new genomic SSR markers. Molecular Breeding, 2017, 37: 93.
[23] SELKOE K A, TOONEN R J. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecology Letters, 2006, 9: 615-629.
[24] 张馨方, 张树航, 李颖, 郭燕, 王广鹏. 基于SSR标记的燕山板栗种质资源遗传多样性分析. 中国农业大学学报, 2020, 25(4): 61-71.
ZHANG X F, ZHANG S H, LI Y, GUO Y, WANG G P. Genetic diversity analysis of chestnut germplasm in Yanshan region based on SSR markers. Journal of China Agricultural University, 2020, 25(4): 61-71. (in Chinese)
[25] 黄武刚, 程丽莉, 周志军, 刘建立. 板栗野生居群遗传多样性研究. 果树学报, 2010, 27(2): 227-232.
HUANG W G, CHENG L L, ZHOU Z J, LIU J L. SSR analysis on genetic diversity of wild Chinese chestnut populations. Journal of Fruit Science, 2010, 27(2): 227-232. (in Chinese)
[26] XING Y, LIU Y, ZHANG Q, NIE X H, SUN Y M, ZHANG Z Y, LI H C, FANG K F, WANG G P, HUANG H W, BISSELING T, CAO Q Q, QIN L. Hybrid de novo genome assembly of Chinese chestnut ().GigaScience, 2019, 8: 1-7.
[27] 郎萍, 黄宏文. 栗属中国特有种居群的遗传多样性及地域差异. 植物学报, 1999, 41(6): 651-657.
LANG P, HUANG H W. Genetic diversity and geographic variation in natural populations of the endemicspecies in China. Acta Botanica Sinica, 1999, 41(6): 651-657. (in Chinese)
[28] MOKAY J. Self-sterility in the Chinese chestnut. Proceedings of the American Society for Floret Science, 1942, 41: 156-160.
[29] PAYNE J A, JAYNES R A, KAYS S J. Chinese chestnut production in the United States: practice, problems, and possible solutions. Economic Botany, 1983, 37: 187-200.
[30] 程丽莉, 胡广隆, 苏淑钗, 黄武刚. 板栗及其近缘种叶绿体SSR遗传多样性分析. 华北农学报, 2015, 30(2): 145-149.
CHENG L L, HU G L, SU S C, HUANG W G. Diversity of cultivated chestnuts and it’s close relative species using chloroplast SSRs. Acta Agriculturae Boreali-Sinica, 2015, 30(2): 145-149. (in Chinese)
[31] WRIGHT S. Evolution in Mendelian populations. Genetics, 1931, 16: 97.
[32] 田华, 康明, 李丽, 姚小洪, 黄宏文. 中国板栗自然居群微卫星(SSR)遗传多样性. 生物多样性, 2009, 17(3): 296-302.
TIAN H, KANG M, LI L, YAO X H, HUANG H W. Genetic diversity in natural populations ofinferred from nuclear SSR markers. Biodiversity Science, 2009, 17(3): 296-302. (in Chinese)
[33] 李颖林. 锥栗野生和栽培群体遗传多样性的SSR分析[D]. 福州: 福建农林大学, 2019.
LI Y L. Genetic diversity of wild and cultivated populations revealed by SSR markers in[D]Fuzhou: Fujian Agriculture and Forestry University, 2019. (in Chinese)
[34] TANAKA T, YAMAMOTO T, SUZUKI M. Genetic diversity ofin Northern Japan assessed by SSR markers. Breeding Science, 2005, 55(3): 271-277.
[35] 韩继成, 刘庆香, 王广鹏, 孔德军, 张新忠. 栗属植物遗传多样性研究进展. 华北农学报, 2006, 21(z2): 129-132.
HAN J C, LIU Q X, WANG G P, KONG D J, ZHANG X Z. The progress of the genetic diversity studies on(Tourn.) L. Acta Agriculturae Boreali-Sinica, 2006, 21(z2): 129-132. (in Chinese)
Genetic Diversity Evaluation ofin China Based on Fluorescently Labeled SSR
NIE XingHua1,ZHENG RuiJie2,ZHAO YongLian3,CAO QingQin1,4,QIN Ling1,4,XING Yu
1College of Plant Science and Technology, Beijing University of Agriculture, Beijing 102206;2Liaoning Economic Forest Research Institute, Dalian 116000, Liaoning;3Chestnut Technology Experiment and Extension Station of Huairou District, Beijing 102206;4Advanced Innovation Center for Forest Molecular Design and Breeding, Beijing 102206
【】The characteristics of genetic diversity, relationships and population structure ofplants in China were analyzed with SSR molecular markers, so as to provide a theoretical basis for variety improvement, germplasm innovation and utilization ofplants. 【】Firstly, 330 SSR molecular markers were screened among 12 chestnut cultivars from different production areas, and 12 pairs of high-quality SSR primers were obtained from them. Secondly, 96 accessions offrom 4 species were detected by high resolution capillary electrophoresis. Finally, Power Marker 3.25, GenAlEx 6.51, FigTree v1.4.3 and Structure 2.3.3 were used to analyze the population genetic diversity of all accessions. 【】A total of 129 alleles were acquired from 96 accessions, and the average variation of each marker was 10.750. The gene diversity (GD) ranged from 0.656 (CmSI0396) to 0.877 (CmSI0930) with an average of 0.800; the observed heterozygosity (Ho) ranged from 0.329 (CmSI0742) to 0.769 (CmSI0702) with an average of 0.615; the expected of heterozygosity (He) ranged from 0.489 (CmSI0742) to 0.789 (CmSI0922), with an average of 0.672; the polymorphic information content (PIC) ranged from 0.586 (CmSI0396) to 0.868 (CmSI0930), with an average of 0.774. Among differentspecies, in terms of number of alleles (Na), number of effective alleles (Ne) and Shannon diversity index,had the highest genetic diversity, followed by, andwas the lowest. According to pairwise population Fst values, the genetic differentiation value ofspecies was 0.077-0.180, showing moderate to high differentiation among the species. Compared with, bothandexhibit relatively high genetic differentiation with Fst values of 0.165 and 0.180, respectively. At the same time, the gene flow (Nm) of theplants was 1.580>1, suggesting frequent gene exchange amongplants, and which therefore reduced the degree of genetic differentiation among the species caused by genetic drift. The results of analysis of molecular variance (AMOVA) showed that the variation mainly occurred within populations, accounting for 73% of the total variation, and the variation among populations made up 27%. The results of UPGMA cluster analysis, principal coordinate analysis and population genetic structure were consistent, and the genetic background of each accessions had obvious inter-species boundaries. Some accessions inherited the genetic information of different ancestor species in generational inheritance. For example, accession 65, 71 and 82 are mixed types, which contain the genetic background ofand, but there is geographic isolation between the two species.plants in the same ecological region existed extensive gene exchange, and there were no complete reproductive isolation. Accession 48 (‘Guangdongaisheng’) had the genetic background of bothand. In terms of geographical distribution, the native place of this accession was in the overlapping ecological area ofand. 【】The 12 pairs of SSR primers screened could accurately assess the genetic diversity ofplants in China. Comprehensive cluster analysis could confirm that the classification ofplants was highly consistent with the inter-species information and there was a certain amount of gene exchange between species.
SSR;; cluster analysis; population genetic structure; population genetic differentiation

10.3864/j.issn.0578-1752.2021.08.013
2020-07-02;
2020-09-15
国家重点研发计划(2018YFD1000605)、国家自然科学基金(31870671)、北京市属高等学校创新团队建设与教师职业发展计划项目(IDHT20180509)
聂兴华,Tel:18813011218;E-mail:niexinghuabua@163.com。通信作者郑瑞杰,Tel:13909844840;E-mail:zhengruijie2006@163.com。通信作者邢宇,Tel:13811529713;E-mail:xingyubua@163.com
(责任编辑 赵伶俐)
猜你喜欢
公民与法治(2022年12期)2023-01-07 09:18:02
小学生作文(低年级适用)(2022年11期)2022-12-02 09:01:10
区域治理(2022年40期)2022-11-27 04:01:54
今日农业(2022年15期)2022-09-20 06:54:16
动漫界·幼教365(小班)(2019年10期)2019-10-28 02:04:20
动漫界·幼教365(大班)(2019年10期)2019-10-28 01:54:09
动漫界·幼教365(中班)(2019年10期)2019-10-28 01:53:17
创新作文(小学版)(2019年10期)2019-09-25 08:12:24
红土地(2018年7期)2018-09-26 03:07:38
小学生作文(中高年级适用)(2018年5期)2018-06-11 01:22:46
