
烃类选择氧化过程中自由基的调控策略与工业氧化应用研究进展
2021-04-20 10:31:54周贤太薛灿纪红兵
化工进展 2021年4期
周贤太,薛灿,纪红兵
(1 中山大学精细化工研究院,化学工程与技术学院,广东珠海519082;2 中山大学精细化工研究院,化学学院,广东广州510275;3 广东石油化工学院化学工程学院,广东茂名525000)
化学工业是我国国民经济的支柱产业,而有机化工又是化学工业的主导产业,包括基本有机化工、精细化工、三大高分子合成材料等。其中有机产品的合成,约90%以上是以三烯(乙烯、丙烯、丁二烯)、三苯(苯、甲苯、二甲苯)、甲烷和乙炔等为基础原料,经催化氧化、磺化、卤化、硝化等有机分子“官能化”反应得到。因此,上述烃类的“官能化”反应已成为有机化工的关键过程,而通过氧化反应获得的化学品又占整个化学品的30%以上,可见烃类选择氧化反应在化学工业中占有极其重要的地位。
氧化反应是许多基本结构化学单元官能化的基础,是现代化学工业生产过程中重要的化学反应。催化氧化可将烷烃、烯烃等烃类化合物转化为醇、醛、酮等含氧化工产品,是提供合成树脂、合成纤维和合成橡胶等大宗化学品以及各类精细化学品的基本石油化工加工工艺。由于碳氢化合物中的C—H键活化能较高,一般所需的反应条件苛刻,而且容易深度氧化成CO2和H2O,选择性难以控制。因此碳氢化合物的选择性氧化是有机分子“官能化”反应中最具挑战性的工艺之一[1]。
目前工业上的氧化过程按所用氧化剂大致可分为两大类:一类为采用化学氧化剂,如重铬酸钾、高锰酸钾、硝酸等;另一类采用绿色氧化剂,如氧气、空气、过氧化氢等。使用化学计量氧化剂存在环境污染、设备腐蚀以及成本昂贵等问题。因此,使用清洁的氧气/空气作为氧化剂替代传统的计量氧化剂,选择合适的催化剂和有效的反应路径,采用对环境危害少或无毒的溶剂,是实现清洁氧化反应的关键技术之一,是实现绿色催化氧化工艺的重要手段,但也一直是当前研究的热点和难点问题[2]。
碳氢化合物的氧化过程涉及碳氢键和氧分子的活化。氧分子的基态是三线态分子,三线态排布的氧分子与碳氢化合物的反应属于能量和自旋禁阻的反应。因此,反应的活化能也较高,通常情况下氧分子表现出非常惰性的化学行为。为了实现氧气直接氧化烃类的反应,需要找到高活性的催化剂来改变氧分子的电子构型,从而达到活化分子氧的目的。目前的碳氢化合物氧化工业普遍存在选择性差、能耗和物耗高且环境不友好、安全隐患高等问题。如乙烯气固相催化环氧化的反应温度高于200℃,丙烯选择性氧化制丙烯酸的反应温度在300℃以上,对二甲苯氧化制对苯二甲酸反应温度高于190℃。
目前我国石油化工行业已进入转型的关键时期,烃类的选择性催化氧化可以提供高档次、高附加值的下游产品,是石化产品结构调整的核心工艺。实现催化选择性的精确控制是绿色化学的重要概念之一,更是工业催化可持续发展的重要驱动力[3]。反应转化率和产物选择性之间的矛盾是目前烃类催化氧化过程的瓶颈。而提高氧化反应效率和选择性的关键在于催化剂的选择,催化剂是氧化工艺的核心。反应转化率和产物选择性之间的矛盾是目前烃类催化氧化过程的瓶颈和催化化学、工业界的国际性难题。烃类的空气氧化均涉及到自由基的过程,这其中的自由基稳定性、传递过程及定向性等都最终影响到产物的选择性。本文从催化烃类氧化技术、自由基机理等角度,对催化剂设计及自由基调控等相关科学问题进行了综述和展望。
1 催化氧化的研究进展
目前大宗含氧有机化学品的工业生产过程中,绝大部分工艺都采用氧气或空气作氧化剂,多数以液相(均相)催化工艺为主。液相体系由于反应物与催化剂同相,不存在固体表面上活性中心性质及分布不均匀的问题,作为活性中心的过渡金属特定活性高、选择性好,同时还具有反应条件不太苛刻、反应比较平稳、易于控制等优点。
典型的液相氧化工业工艺有:异丁烷氧化制备叔丁基过氧化氢和异丙苯氧化制备异丙苯过氧化氢,这都是两步法制备环氧丙烷的重要一步。环己烷氧化制备环己醇和环己酮(KA 油)是工业生产己内酰胺和己二酸的前端工艺。甲苯和对二甲苯选择性氧化生成芳酸化合物,芳酸是作为生产聚酯纤维、薄膜、医药、有机颜料和农药等重要工业原料的前体,需求量逐年增长。常见的烃类液相选择性氧化工艺及催化体系如表1所示。
从表1可以看出,由于烃类选择性氧化的条件都比较苛刻,需要较高温度和压力下通过催化过程采用强制氧化过程来使得碳氢键均裂产生自由基而实现。另外,所用的催化剂以钴盐和锰盐为主,主要是乙酸钴、乙酸锰、NH4Br或四溴乙烷组成的混合催化剂(MC 催化剂)。该催化体系广泛应用于芳烃侧链化合物及其他烃类物质的选择性氧化。但该工艺使用具有强腐蚀性的溴化物,存在环境和安全等方面的问题。
催化剂的开发是发展绿色催化氧化工艺的关键,国内外研究者开发了许多用于催化氧化反应的新型高效催化剂,包括均相催化剂、非均相催化剂、仿生催化剂等。均相催化剂主要包括改进的乙酸钴/乙酸锰催化剂、过渡金属化合物、自由基引发剂等,非均相催化剂主要包括分子筛、杂多酸、金属有机框架化合物、金属纳米粒子等,仿生催化剂主要包括金属卟啉和其他仿酶催化剂等。
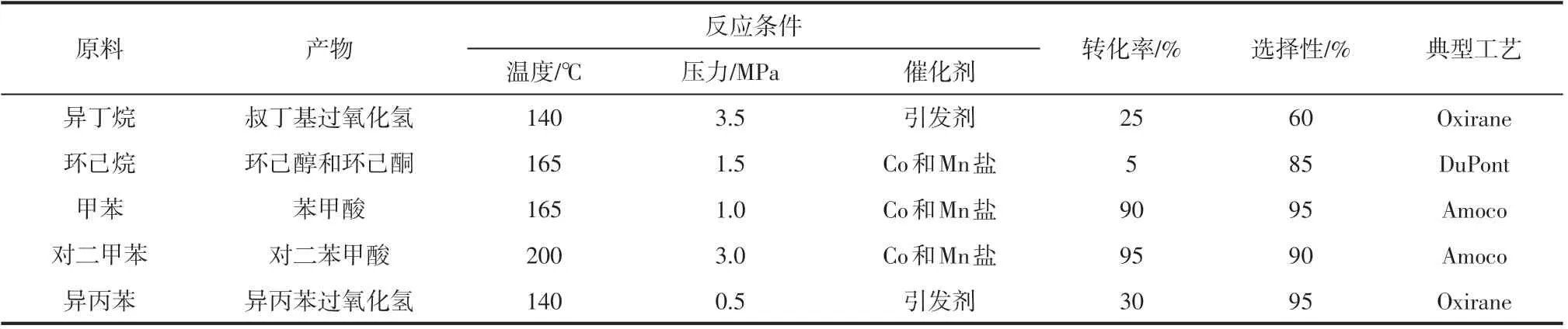
表1 典型的烃类液相选择性氧化工艺及反应条件
1.1 均相催化剂
过渡金属无机盐或与各种配体形成的有机金属配合物是目前常用的均相催化剂[4-7],催化机理一般可以归类为直接氧化底物,或者与活性氧反应生成高价金属氧物种,通过高价活性物种将氧传递给底物,实现底物的氧化。
Murahashi等[8]报道了一种使用铜金属盐催化氧化烷烃的方法。该方法使用乙醛作为助剂,在乙腈/二氯甲烷混合溶剂中,70℃下能高效实现烷烃的催化氧化(图1所示)。以环己烷为模型反应物,环己醇和环己酮产率分别能达到22%和29%。值得注意的是,使用其他的铜盐,例如CuCl2、CuBr2、CuOAc、CuCN和[Cu(CH3CN)4](PF6)等皆具有相似的催化活性,即使是零价的铜粉末也能催化该反应。推测可能是因为铜盐在乙腈溶剂体系中都会转化成亚铜盐形式的配合物[Cu(CH3CN)m(X)](X=Cl,Br,OAc等)。
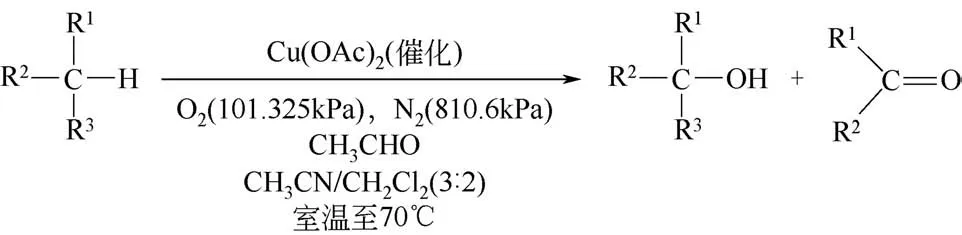
图1 乙酸铜催化烷烃C—H键氧化
Kantam 等[9]对工业甲苯氧化的MC 催化剂进行改进,用NaBr代替ZnBr2,降低了催化体系中的Br量,并考察了分别以乙酸和苯甲酸作为溶剂对甲苯选择性氧化反应的影响。实验发现,不同溶剂对甲苯选择性氧化反应的产物分布有显著影响。以乙酸钴为溶剂、甲苯转化率为38.3%时,苯甲醛收率可达17.3%。钒酸盐被发现是有效催化烷烃羟基化的催化剂,进一步通过机理研究发现,五价的钒酸盐通过还原变成具有催化活性的六价物种,该物种加氧后形成钒过氧化合物,钒金属过氧化合物通过自由基均裂生成羟基自由基和钒氧自由基。羟基自由基是催化氧化体系的活性基团,与烷基自由基结合生成醇类化合物[10]。另外,由Barton教授提出的Gif催化体系,其催化剂组成包括乙酸、FeCl3、Zn、吡啶等,该催化剂可在室温或低温的条件下将烷烃或烯烃中的仲碳氧化生成酮[11]。在催化环己烷氧化中,环己烷的转化率一般在20%~30%,环己酮几乎是唯一产物,没有过度氧化现象发生[12-13]。
自由基的引发对烃类选择性氧化过程至关重要,Ishii教授[14]发现N-羟基邻苯二甲酰胺(NHPI)及其衍生物在钴盐作用下,可高效催化乙烷、异丁烷及环己烷等烃类氧化生成醇和酮。大多数以NHPI 为催化剂的反应需要过渡金属盐(例如Co[15]、Mn[16]、Cu[17]和Fe[18])或者非均相催化剂[19-22]共同催化反应,也可以通过加入过氧化物[23]、单电子转移试剂、光照[24-25]或者加热[26-27]的方式活化NHPI而引发反应。NHPI独特的反应活性是源于其O—H 键解离能(88.17kcal/mol,1cal=4.1840J)大于或接近大多数有机分子的C—H 键解离能(85~100kcal/mol),因而能够从底物上夺取氢,使其转化为活泼的自由基衍生物,达到活化底物分子的效果(图2)[28]。相比之下,2,2,6,6-四甲基哌啶氧化物(TEMOH)的O—H 键的键解离能仅69.6kcal/mol,较大的能量差异很难催化烃类C—H键活化,常用作醇类化合物氧化的催化剂[29]。
NHPI 及其衍生物催化碳氢化合物氧化的过程如图3所示。目前普遍认为此类有机催化剂,无论有无金属催化剂参与,该反应的起始过程都是将NHPI 转化为邻苯二甲酰亚胺N—O 自由基(PINO)。PINO 能够从烷烃上夺氢,将其活化为烃基自由基,并重新转化为NHPI,实现氮氧自由基催化剂的循环。与此同时,活泼的烃基自由基与分子氧迅速反应生成烃基过氧自由基,过氧自由基可以从NHPI上夺氢生成PINO(路径a)。在共催化体系中,金属催化剂在高价氧化态也能促进NHPI 转化为PINO(路径b)。烃基过氧化物受热或金属催化剂作用下O—O键分解得到氧化物。

图2 常见有机催化剂和C—H键的键解离能
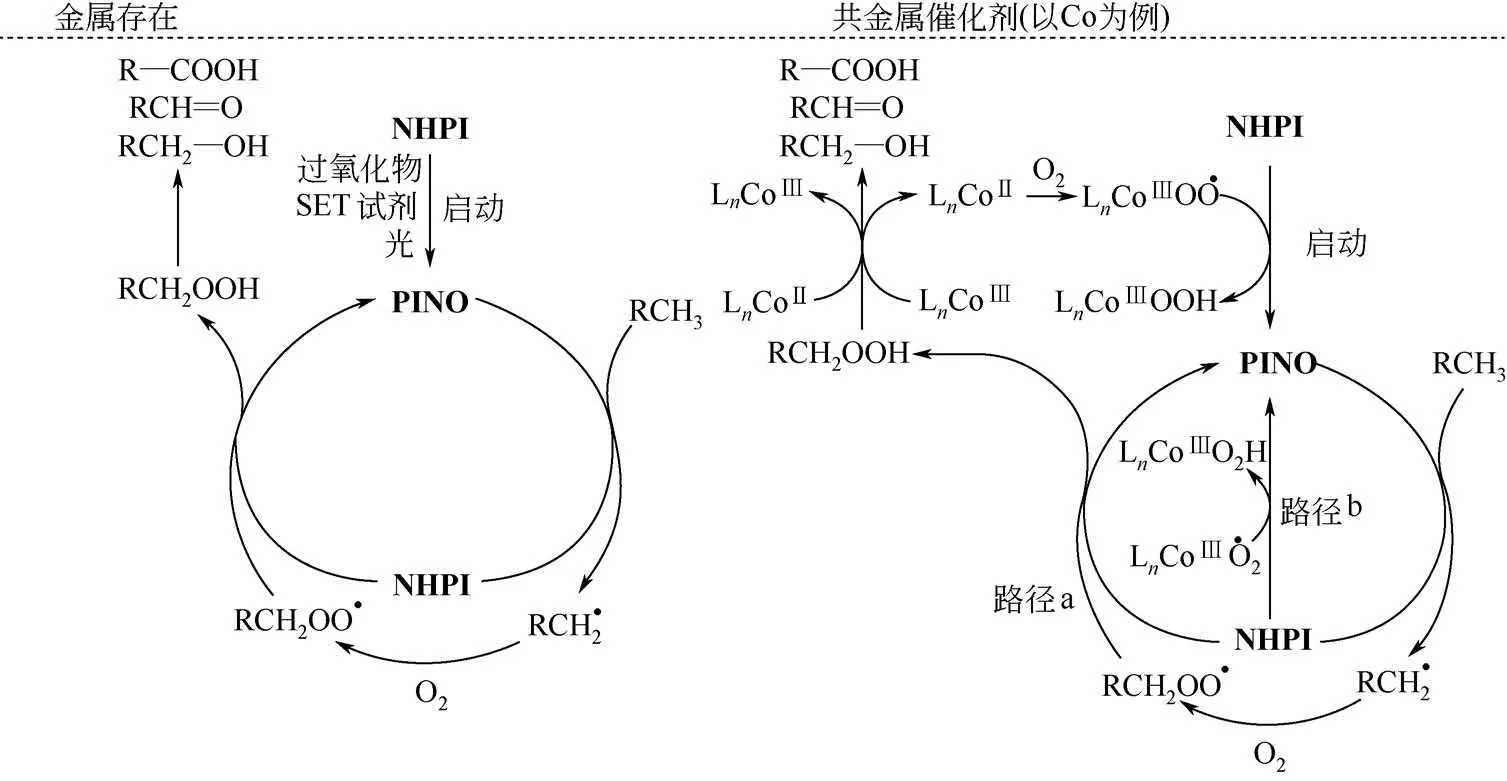
图3 NHPI参与下的C—H键氧化机理
在甲苯氧化反应中,生成的苯甲醛比甲苯更容易发生氧化,在反应过程中苯甲醛会过度氧化苯甲酸,这是甲基芳烃化合物在氧化过程中常发生的问题。Gaster 等[30]使用六氟异丙醇(HFIP)作为反应溶剂,实现了甲苯高选择性催化转化为苯甲醛,如图4。他们推测可能是因为HFIP 与醛类化合物会形成强氢键作,抑制了醛基上的C—H键的过度氧化[31-32],并且底物同时有多个甲基取代基的情况下,利用该反应体系也可以实现单一甲基选择性氧化为醛基。
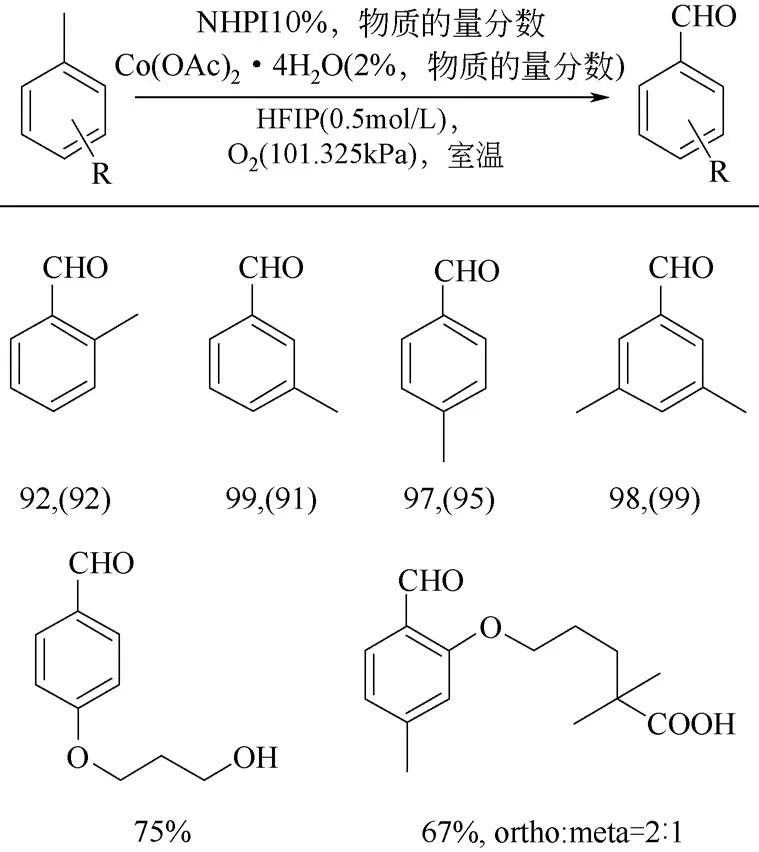
图4 Co/NHPI催化芳烃侧链的选择性氧化
虽然NHPI在C(sp3)—H键选择性氧化中表现优异,但是NHPI生成PINO往往需要高温条件(80℃以上),而该条件下PINO易分解成邻苯二胺、邻苯二酸酐或形成不活泼的三聚物[33-34]。为了增强NHPI的稳定性,人们研究将NHPI 固定在载体上,同时便于回收利用。
1.2 非均相催化剂
在非均相催化反应中,催化剂的相和反应底物的相不同,因此相较于均相催化剂更容易分离,但是其活性一般比均相催化剂低。从结构上看,非均相催化剂可以分为活性组分、助催化剂和载体。其中,在催化反应中起到催化活性作用的决定性物质是活性组分。它可以是一种物质也可以是多种物质组成。助催化是添加到催化剂中的少量组成成分,起辅助催化剂活性组分的作用,一般情况下自身无催化活性或活性较低,但是能够通过改变催化剂的化学性质、中心离子价态、表面结构和物理性质等,从而增强催化剂的活性和稳定性,是不可或缺的一部分。载体主要起承载活性组分和助催化剂的作用,其物理结构和性质在某种程度上对催化剂活性具有决定性的影响。金属氧化物、金属有机框架化合物和各种碳材料(如石墨烯、碳纳米管)是常用的多相催化剂材料[35-39]。
金属-有机框架材料(MOFs)是过渡金属离子与有机配体通过自组装的方式形成的一类具有周期性结构的晶体多孔材料。虽然关于MOFs材料的报道很多,但是关于其在催化领域的应用相对较少[40-41]。MOFs 材料的一大优点是制备简单,容易对有机结构框架进行修饰,可以对催化剂的结构和活性进行调控。此外,由于其高比表面积和孔隙率,它们是金属纳米粒子的优良载体。Li等[42]报道了一种将Au-Pd 双金属合金纳米粒子负载在MIL-101上的催化剂,在催化环己烷氧化方面具有优异的性能,反应过程如图5。MIL-101 是由对苯二甲酸和铬盐形成的一种沸石型金属有机框架化合物[43]。环己烷的选择性氧化一直是工业上的一个难题,很难做到在保持高转化率的情况下能高选择性的得到KA 油。值得注意的是,使用Au-Pd/MIL-101 催化剂在环己烷转化率达到30%以上时KA 油仍然具有很高的选择性(>80%)。这种高选择性可能是由于双金属Au-Pd 纳米粒子的协同合金化效应。
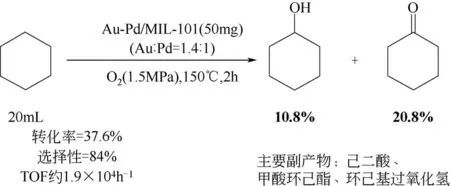
图5 Au-Pd/MIL-101催化氧化环己烷
Unnarkat 等[44]报道了在无溶剂条件下用分子氧在钴钼氧化物催化剂上氧化环己烷。以三种不同Co∶Mo 物质的量之比(1∶1、1∶2、2∶1)的柠檬酸盐和不同的氧化物制备催化剂,并进行了比较研究。所有催化剂均表现出显著的活性和选择性,与其他催化剂相比,1∶1 配比的CoMoO4的转化率最高,转化率为7.38%,KA油的选择性为94.3%。
Mistri 等[45]首次报道的铜离子取代铝酸盐中的MAl2O4(M=Mg,Mn,Fe,Ni,Zn),尖晶石CuxM1-xAl2O4(x=0.03, 0.05) 通过单步溶剂燃烧法合成。Cu0.03Fe0.97Al2O4在乙腈中,以过氧化氢为氧化剂氧化环己烷的反应活性(转化率为92%,选择性为99%)明显高于其他含铜类似物。而浸渍催化剂中含有分散良好的CuO 微晶。循环作用(重复三次)对铜离子取代的铁铝酸盐活性几乎没有影响。相比之下,由于分散的CuO 微晶存在,类似的浸渍催化剂在连续循环中表现出相当大的活性损失[45]。杂多化合物催化剂催化环己烷氧化时,一般需要一定量的自由基引发剂或还原剂,以及降低反应温度,才能抑制反应的进一步深度氧化。
1.3 仿生催化剂
金属卟啉配合物是细胞色素P-450单加氧酶的有效模拟物,相对化学催化剂而言,金属卟啉仿生催化剂能在较温和的条件下实现对分子氧的活化[46-49]。而四苯基金属卟啉是最简单的金属卟啉仿生催化剂,易于合成且结构非常简单,但催化性能较差,稳定性不太好,容易分解。因此,在仿生催化氧化体系中会对金属卟啉催化剂进行基团的改性,以提高其催化性能和选择性。
目前还尚未有金属卟啉仿生催化乙烯、乙烷及丙烷选择性氧化方面的相关报道。而针对丙烯,由于其结构中具有活泼α氢,α氢的性质比双键更活泼,因此催化丙烯选择性环氧化的研究是非常具有挑战性的[50-52]。环氧丙烷又是一类用途极广的有机合成中间体,广泛应用于石油化工、精细化工和有机合成等领域。本文作者课题组[53]通过在仿生催化体系中加入递氢体后,以MnTPPCl(1×10-4mol/L)为催化剂、乙腈为溶剂、80℃的反应条件下获得了66%的丙烯转化率和89%的PO 选择性,实现了金属卟啉仿生催化丙烯环氧化过程中转化率和选择性的有效统一。通过原位技术手段、量子化学计算等方法的机理研究表明,其作用机制为过酸氧化和高价活性金属物种共同作用(如图6)。
对于金属卟啉催化低碳烷烃如碳四、碳五烷烃的氧化,报道的例子不是很多。早期Ellis等[54]研究了氯代苯基金属卟啉催化氧化异丁烷制备叔丁醇,研究中发现,当金属卟啉外环上有取代基时,可以提高催化剂的催化性能和抗氧化性能,叔丁醇的选择性达到90%以上,其中催化剂活性转化数高于13000。随后Lyons等[55]报道了在80℃的条件下,对比了不同金属离子的催化效果,用取代基相同但金属离子不同的卟啉为催化剂,乙苯为溶剂,氧气氧化异丁烷氧化制备叔丁醇,研究发现铁卟啉性能最好,活性高于铬卟啉和锰卟啉。由此他们认为当金属还原电位越高时,相应的金属卟啉催化性能往往越好。
环己烷氧化得到的主要产物环己酮是生产己内酰胺和己二酸的基本原料。湖南大学郭灿城等[56-57]开展了系列金属卟啉催化环己烷氧化制备环己酮、环己醇(KA 油)的仿生催化氧化体系,并建立了中试装置,进行了连续的实验。较现有生产工艺,金属卟啉仿生催化环己酮空气氧化可实现转化率提高将近一倍,环己酮选择性可以达到90%。而简单结构的铁卟啉可以实现环己烷一步氧化到己二酸,己二酸的收率可高达21.4%,催化剂的活性转换数高达25000[58]。以四(4-氯苯基)卟啉氯化锰(Ⅲ)为催化剂,苯甲酸为助催化剂,O2为氧化剂,140℃实现了环己烷的催化氧化,高选择性合成己二酸和戊二酸,无溶剂条件下环己烷转化率达到16.4%,己二酸和戊二酸的总选择性达到56%[59]。
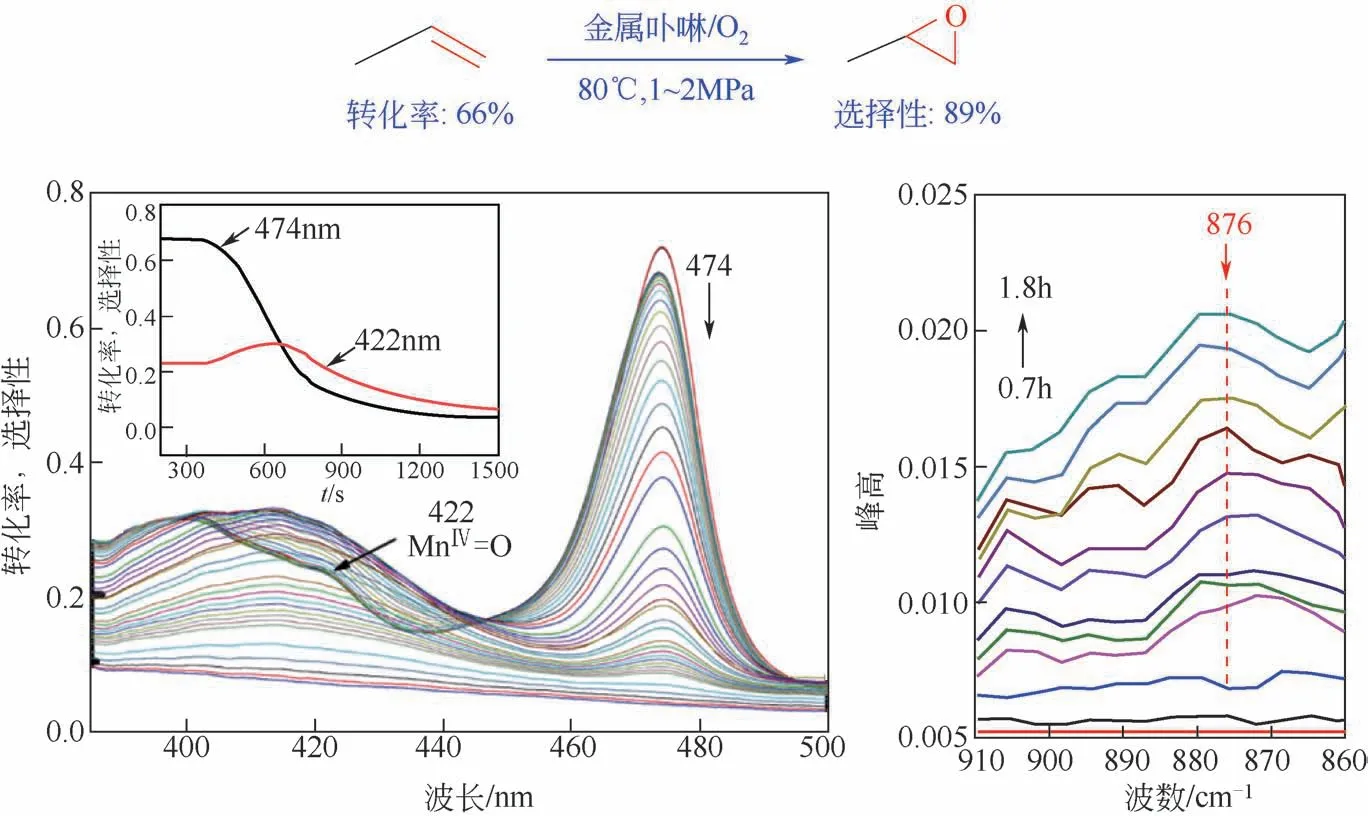
图6 金属卟啉仿生催化丙烯液相选择性环氧化的性能与原位紫外表征
同时以简单的卟啉钴(Ⅱ)和卟啉铜(Ⅱ)组成二元催化剂,O2为氧化剂,120℃实现了环烷烃的催化氧化,高选择性合成环烷醇和环烷酮。以四(2-氯苯基)卟啉钴(Ⅱ)和T(4-氯苯基)卟啉铜(Ⅱ)为催化剂,无溶剂条件下环己烷的单程转化率达到4.41%,环烷醇和环烷酮的总选择性达到97%。环庚烷、环辛烷和环十二烷为底物,醇酮选择性均在99%以上[60]。
PoŁtowicz 等[61]研究了以氧气为氧化剂,Mn、Co、Fe 卟啉为催化剂催化氧化环辛烷,实验结果表明金属卟啉Mn(TPFP)Cl 的活性最好,产物主要是以环辛酮为主,其中环辛酮和环辛醇的收率分别为17.2%、3.2%,催化剂活性转化数高达23584。Haber 等[62]在环辛烷的催化氧化反应中,当在催化反应中用以—Cl为轴向配体,吡咯β位用—Br取代的Mn(TPFPβBr8P)Cl 为催化剂时,环辛酮收率高达20%,活性转化数44242。但是这一类催化剂的合成工艺复杂,成本昂贵。
在金属卟啉催化芳烃化合物的氧化方面,主要有甲苯、乙苯、对二甲苯等的氧化。Huang 等[63-64]在温度165℃、压力0.8MPa、0.04m3/h 的空气流速连续通氧的条件下,采用钴卟啉作为催化剂对甲苯进行催化氧化,转化率可以达到8.9%,同时醛醇选择性可以达到近60%,催化剂的转化数约为25000,反应条件为工业可接受的温和条件,且反应具有比较高的选择性。进一步提高反应温度和压力后(200℃,压力1.2MPa),反应转化率可达14.7%,但是醛醇的选择性下降到40%,产物苯甲醛和苯甲醇的收率仅为5.9%。
佘远斌教授团队[65]以简单的金属卟啉和NHPI组成二元催化剂、O2为氧化剂、120℃实现了芳烃苄位伯C—H键的催化氧化,高选择性合成芳烃羧酸。以四(2-甲氧基苯基)卟啉钴(Ⅱ)和NHPI 为催化剂,无溶剂条件下甲苯转化率达到35%,苯甲酸选择性达到92%。该催化体系对其他芳烃苄位伯C—H 键的催化氧化也具有很好的底物普适性。该团队进而以简单的金属卟啉为催化剂,氧气为氧化剂,温和条件下实现了芳烃苄位仲C—H键的催化氧化,高选择性地合成芳香酮。以四(2,3,6-三氯苯基)卟啉钴(Ⅱ)为催化剂、120℃、无溶剂条件下4-硝基乙苯转化率达到47%,4-硝基苯乙酮选择性达到87%。该催化体系对其他芳烃苄位仲C—H键的催化氧化也具有很好的底物普适性[66]。
佘远斌教授团队[67]在温和条件下,以简单结构的金属卟啉为催化剂,氧气为氧化剂,实现了芳烃苄位叔C—H键的催化氧化直接合成苄位叔醇。以四(2,3,6-三氯苯基)卟啉锰(Ⅱ)为催化剂,70℃、无溶剂条件下异丙苯转化率达到57%,2-苯基-2-丙醇选择性达到70%。该催化体系对其他芳烃苄位叔C—H键的催化氧化也具有很好的底物普适性。
2 催化氧化的自由基研究进展
2.1 催化氧化的自由基机理
催化氧化涉及到碳氢化合物底物和分子氧的活化,碳氢化合物和分子氧的结构特征决定二者的反应为自旋禁阻反应。一般地,氧气需要被金属催化剂活化,变成单线态形式的M/O2物种,或者C—H键被自由基或强碱活化,变成碳自由基或碳负离子中间体,然后二者之间才能发生C—H键的羟基化反应。
Hong 等[68]用Co/ZSM-5 催化环己烷选择性氧化制备KA 油,并提出了催化环己烷氧化的机理如式(1)~式(6),在反应条件下环己烷脱氢环己烷自由基(C6H11·),该自由基迅速与分子氧反应生成环己基过氧化氢自由基(C6H11OO·)。环己基过氧化氢自由基(C6H11OO·)可以进一步与一个环己烷分子(C6H12)反应生成环己基过氧化物C6H11OOH(CHHP),并生成另一个环己烷自由基(C6H11·)生成。催化剂Co3+与环己基过氧化物C6H11OOH 生成环己基过氧化氢自由基(C6H11OO·)。Co2+与C6H11OOH 生成环己基烷氧基自由基(C6H11O·)。环己基烷氧基自由基(C6H11O·)结合环己烷分子的氢后生成环己醇(C6H11OH)和另一个自由基(C6H11·)。两个自由基(C6H11OO·)重排生成环己醇(C6H11OH)和环己酮(C6H10O)。
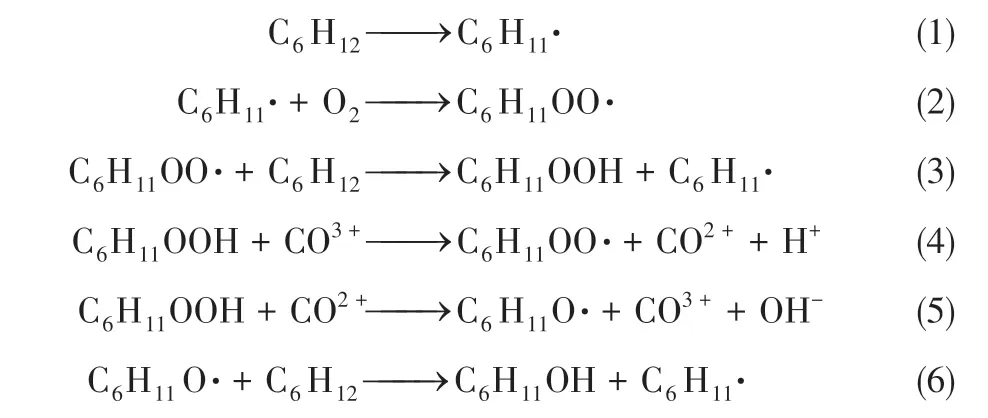
催化烃类选择性氧化机理中的关键步骤是自由基的引发,因为C—H键的键能较高,所以所需的条件较苛刻。常规碳氢化合物的空气氧化主要通过氧化型金属盐类催化或无催化的气-液反应,反应经历了称为Haber-Weiss 循环的以活化碳氢键均裂为链引发的自由基反应[69]。金属卟啉作为仿生催化剂,在催化烃类氧化中,通过催化剂来实现氧气的活化,形成PorM-OO·,进而引发底物形成自由基。如钴卟啉催化环烷烃的选择性氧化的过程中,不同取代基的金属卟啉其活性有差异,结果发现含有吸电子基团越多的催化活性越高[70]。通过密度泛函理论(DFT)计算表明吸电子取代基越多的钴卟啉越容易形成Por-Co-O2中间体[71-72]。以环辛烷为模型底物,该研究提出了一个钴卟啉催化环辛烷氧化的自由基链式反应机制,如式(7)~式(15)。
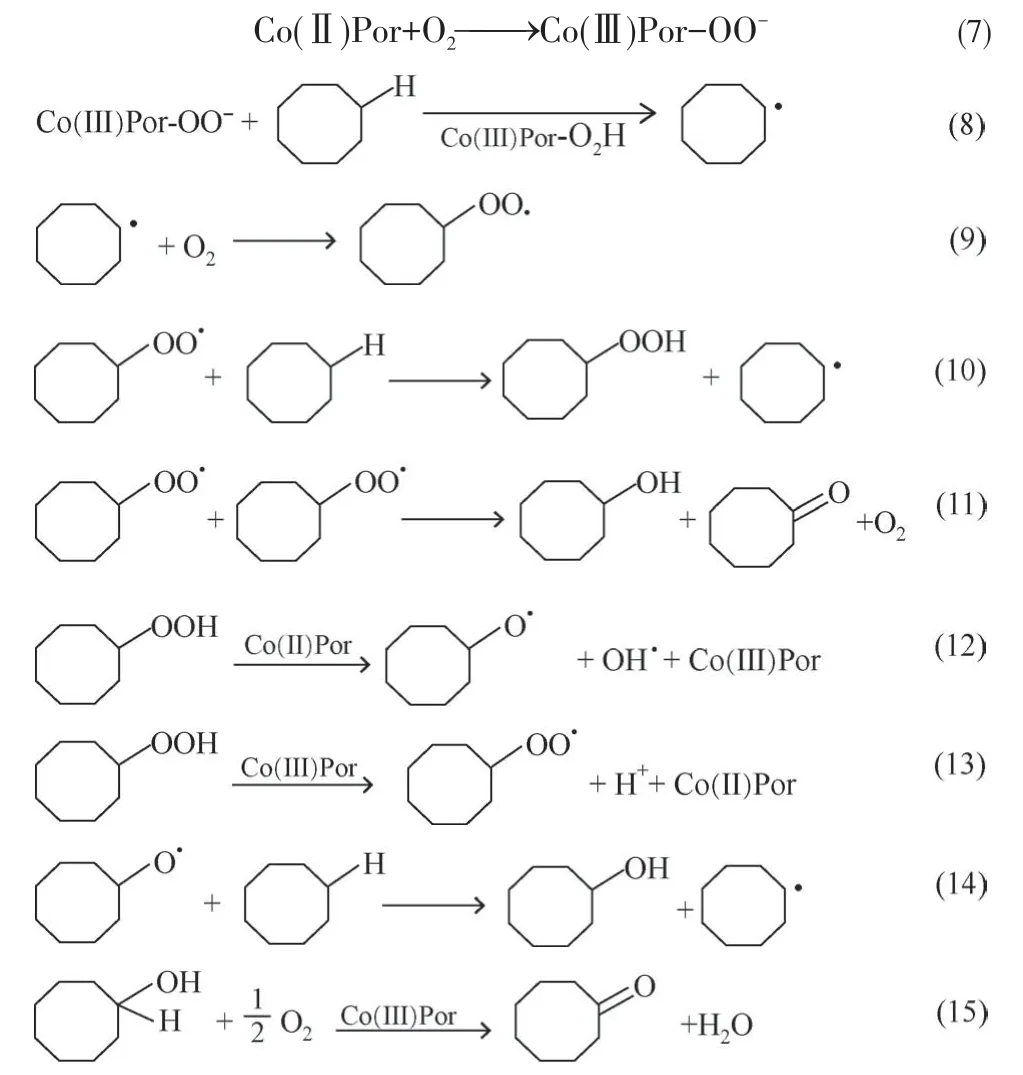
金属卟啉催化剂催化烷烃的氧化过程中,普遍被认可的机理是类似细胞色素P-450单充氧酶的催化机理,即反应经历以活化分子氧为特征的高价铁氧正离子自由基提取碳氢化合物氢原子的笼内自由基反应历程[73]。该反应首先由钴卟啉活化分子氧,从而形成超氧化物物种[式(7)]。下一步,后者从环辛烷上夺氢,形成环烷基自由基R·[式(8)],然后环烷基自由基R·和分子氧耦合,得到环烷基过氧自由基ROO·[式(9)]。环烷基过氧自由基ROO·可进一步与另一分子环辛烷反应生成环烷基过氧化氢ROOH 和环烷基自由基R·,从而实现链式循环[式(10)]。在式(11)中,在链终止反应中ROO·的重排则可以形成环酮和环醇衍生物。二价钴卟啉可以还原O—O 键进而得到环烷氧基自由基RO·和氢氧根负离子[式(12)],而三价钴卟啉可以继续氧化环烷基过氧化氢ROOH 形成环烷基过氧自由基ROO·[式(13)]。环辛醇的另一个来源是在式(12)中产生的环烷氧基自由基RO·夺氢产物[式(14)]。在上述步骤中产生的环辛醇也可以在催化剂的存在下进一步氧化为环辛酮[式(15)]。
由此可知,不同的催化体系所导致经历的自由基历程有所区别。不同的自由基历程所对应的反应条件、产物选择性必然有较大差异。因此,如何通过自由基的调控,实现温和条件下自由基的引发和传递是解决氧化反应总转化率和选择性之间矛盾的关键。
2.2 自由基的调控
一般而言,金属催化剂对氧气直接活化所需的条件比较苛刻。为了实现温和条件下氧气的活化,通过模拟生物酶催化反应的历程,提出了引入递氢体调变氧气的活化历程。在构筑的仿生催化体系中,以醛类物质、烯烃及烷烃等作为递氢体,在金属卟啉的作用下脱氢生成自由基,通过自由基的调变完成了温和条件下氧气的活化。与大多传统化学氧化中氧气的活化方式不同,递氢体的引入由于改变了直接由催化剂活化氧气的途径,才使得温和条件下的氧气活化得以顺利实现。同时,递氢体结构的调变也使自由基的生成速率、氧气活化过程也得到了调控(如图7)。
本文作者课题组[74]在金属卟啉仿生催化二苯甲烷直接氧化的研究中发现在较苛刻的反应条件(150℃、2.0MPa)下,反应8h 后二苯甲烷的转化率仅为7.6%。而当在体系中加入环己烯作为递氢体后,即使将反应温度降低到110℃,反应压力降为1.0MPa时,二苯甲烷的转化率可达到42.5%,为之前的6倍。进一步的机理研究发现,加入环己烯后改变了氧气的活化模式,环己烯失去α-H 后形成自由基,通过该活性自由基来实现氧气的活化,该自由基与氧气结合,生成过氧自由基,过氧自由基进而与金属卟啉作用,生成烷氧自由基和高价金属卟啉氧代物。烷氧自由基可以使烷烃二苯甲烷脱氢,形成烷基自由基,高价金属氧代物可将氧直接传给底物,实现烷烃的羟基化,生成二苯甲醇或二苯甲酮。
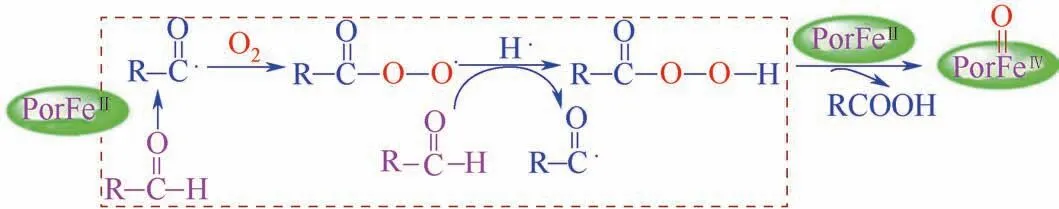
图7 铁卟啉仿生催化的氧气活化过程
在金属卟啉催化甲苯的液相氧化中,加入在以环己烯为递氢体的仿生催化甲苯氧化反应中,以MnTPP 作为催化剂、乙腈作溶剂,通过优化,获得了优化的反应条件,即反应温度160℃,压力1.2MPa,催化剂用量为1.0×10-3mmol,反应4h 后,甲苯转化率达到12.5%。利用原位紫外和原位电子顺磁共振等表征手段证明了该反应机理,环己烯易失去α-H 生成活性自由基,该活性自由基易活化分子氧生成过氧化物,进一步与金属卟啉相互作用生成具有高活性的金属-氧高价化合物,从而促进甲苯催化氧化反应的进行。具体来说,环己烯失去α-H 生成的活性自由基易于活化分子氧,与O2结合生成过氧自由基,在体系内夺得一个质子,生成过氧化物。过氧化物在金属卟啉的作用下,分解成氧自由基,同时金属卟啉被氧化成高价氧化物。该高价氧化物和过氧化物可进一步活化底物甲苯生成苄基自由基,苄基自由基与氧气反应生成苄基过氧自由基,苄基过氧自由基夺取体系中的一个质子,生成苄基过氧化物。苄基过氧化物进一步在金属卟啉的作用下,分解产生苄基氧自由基,最终生成苯甲醇。另一条路径,由苄基过氧化物脱去一分子H2O 生成苯甲醛,并进一步氧化生成苯甲酸[75],如图8所示。
在仿生催化氧化体系的自由基调控研究中,原位电子顺磁共振表征是重要的表征方法。通过设计合理的实验以及相关的模拟方法,可以探究仿生催化氧化体系中自由基的形成过程及形成规律,探究自由基的稳定性,并对所有的自由基进行归属。
如在锰卟啉(MnTPPCl)驱动叔碳基自由基活化O2及催化氧化过程中电子转移机制的研究中,利用原位电子顺磁共振表征技术,重点分析了体系中自由基传递过程对烃自由基形成的促进作用机理、烃自由基形成的活化能影响等(如图9)。

图8 锰卟啉仿生催化甲苯-环己烯共氧化
在常压和80℃下,环氧化物收率达到59%~99%,酮收率达到50%~96%。以二苯甲烷氧化为模型反应,共底物的特异性研究显示苯环有助于叔碳基自由基的稳定,并且共底物的给电子基团不利于底物氧化。米氏动力学显示MnTPPCl/异丙苯催化体系是一个类酶动力学过程。通过原位紫外可见吸收光谱和原位电子顺磁共振,研究了氧化反应过程中的电子转移机制。结果显示:共底物异丙苯氧化是一个自由基反应,中间产物异丙苯过氧化氢与MnTPPCl 相互作用,通过O—O 键异裂得到高价锰氧自由基正离子(MnⅣ==O⋅+);它进而夺取底物二苯甲烷的氢原子得到烷基自由基,该自由基倾向于从自由基笼中逃离,与残余的MnⅣ==O⋅+通过自由基偶联得到二苯甲酮[76]。
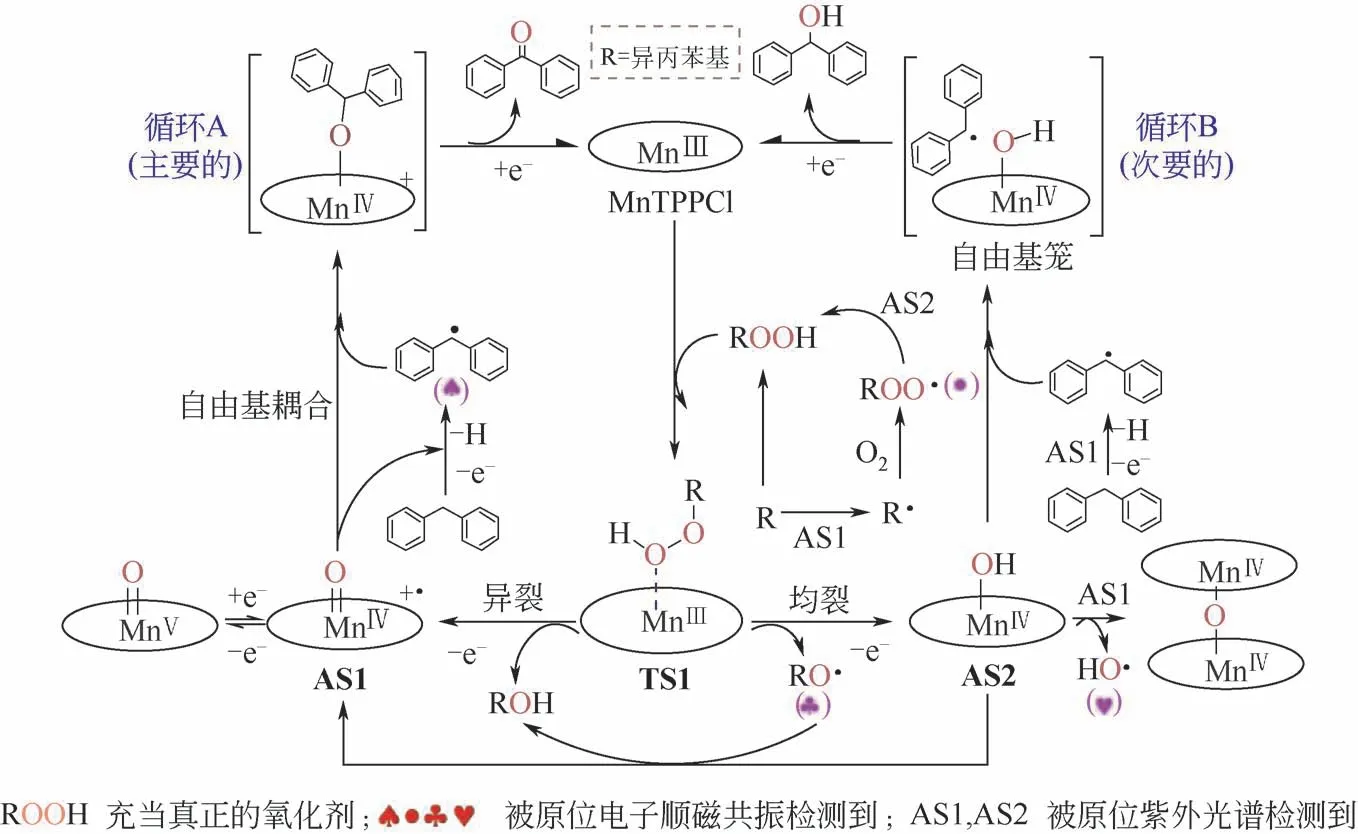
图9 仿生催化二苯甲烷氧化中的自由基传递和可能机理
2.3 仿生催化氧化法制备ε-己内酯的连续化工业级中试示范
ε-己内酯是一种重要的高分子聚合单体,作为新型聚酯单体,被广泛应用于合成各种不同用途的聚己内酯和共混改性树脂。目前全球仅有瑞典Perstorp、日本大赛璐和美国BASF 三家公司生产。在各自的生产工艺中,都是通过Baeyer-Villiger 氧化反应来生产ε-己内酯,采用的氧化剂通常是有机过酸如过氧甲酸、过氧乙酸、过氧苯甲酸等,使用该类氧化剂不仅费用高,而且在实际操作中存在着诸多安全隐患,特别是其合成前期过氧酸的浓缩以及后续纯化过程所产生的浓度过高、易爆的过氧化物使得该工艺的实际应用受到限制。
本文作者课题组在研究仿生催化氧化的基础研究之上,解决了复杂反应动力学、高精度要求的反应器设计、产品分离难度大等技术难题,研究了氧气在反应溶液中的扩散系数、构建了包含传质因素在内的反应动力学模型,设计了100L 的中试反应器,同时提出了两段精馏的分离工艺;建立了仿生催化氧化的中试装置(300t/a 的规模),实现了仿生催化环己酮氧化制备ε-己内酯的连续化中试工艺。目前已在山东淄博试车成功,环己酮的转化率为40%,ε-己内酯选择性为86%。研究团队攻克了温和条件下实现氧气活化的科学难题,通过自由基的调控和传递控制,实现了工艺的高效性、高选择性及高安全性。该工艺技术具有安全、绿色、低能耗等特点,完全具有自主知识产权,填补了国内外相关技术的空白(如图10)。

图10 仿生催化氧化制备ε-己内酯300t/a的中试现场及装置
3 结语与展望
本文系统总结了目前催化氧化制备大宗含氧化学品及高附加值精细化学品方面的研究进展,并详细综述了仿生催化烃类选择性氧化的研究现状,围绕催化氧化的自由基机理,介绍了温和条件下氧气的活化机制、活性氧的传递规律、自由基稳定性和定向性调控机制等方面的研究现状,介绍了仿生催化氧化制备ε-己内酯和环氧环己烷的工业化示例。
未来,在催化烃类选择性氧化的自由基调控方面,重点要建立涉及自由基的传递速率模型和调控方法,发展基于自由基传递速率模型的科学计算方法,并建立基于自由基调控理论的工业应用示范装置。通过催化工艺的革新,引领催化氧化的产业技术革命,有助于推进我国在烃类仿生催化氧化基础研究进入国际前沿,促进我国石油和化学工业通过自主技术创新以实现跨越式发展,巩固和发展我国在碳氢化合物仿生催化氧化工业应用方面的领先地位和产业竞争力。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12 02:07:48
云南化工(2021年6期)2021-12-21 07:30:56
化工管理(2021年7期)2021-05-13 00:45:22
科学(2020年2期)2020-08-24 07:57:00
环境保护与循环经济(2020年4期)2020-06-08 10:43:48
中国特种设备安全(2019年1期)2019-03-13 01:06:28
中国调味品(2017年2期)2017-03-20 16:18:13
中学化学(2015年2期)2015-06-05 07:18:13
生物技术通报(2015年1期)2015-04-10 16:15:19
无机化学学报(2014年3期)2014-02-28 17:30:58
