
腓骨肌萎缩综合征患者MFN2基因突变检测的研究
2021-04-15 04:51刘红仙段丽芬刘晓梅王惠萍王晓辉褚嘉祐杨昭庆
国际检验医学杂志 2021年7期
刘红仙,段丽芬,刘晓梅,王惠萍,王晓辉,褚嘉祐,孙 浩,杨昭庆△
1.中国医学科学院/北京协和医学院医学生物学研究所,云南昆明 650118;2.云南省昆明市儿童医院,云南昆明 650228
腓骨肌萎缩综合征(CMT)是一类外周神经退行性遗传病,具有显著的临床表型和遗传异质性[1]。CMT在普通人群中的患病率约为1/3 300[2]。CMT一般最早的症状是由足部肌肉萎缩引起,常导致足部畸形,例如高足弓、平足或卷曲脚趾,下肢腿部难以弯曲。CMT有明显的临床异质性,常难以明确认知和诊断。CMT通常在青春期或成年早期表现出明显症状,但是发病时间可能是儿童早期到成年后的任何时候。即使是同一家庭的成员,CMT症状的严重程度和发病年龄也有所不同。基于对CMT发生发展的认知及临床诊治的现状,本研究拟利用全外显子组测序(WES)对1个疑似CMT核心家系的致病突变进行鉴定和遗传学分析,旨在探讨引起突变的原因,进而证实该突变的致病性,为临床诊治提供参考。
1 资料与方法
1.1一般资料 1个核心家系来自云南省,包括先证者及其父亲、母亲和妹妹。先证者,男,10岁,表现出足部畸形,发病于3岁左右;先证者父亲未见四肢肌张力及功能等表型异常;先证者母亲自述8~9岁逐渐出现行走姿势异常及四肢肌肉萎缩,后出现下肢瘫痪,现仅能独坐,不能站立和行走,四肢纤细、消瘦,肌容积明显减少;先证者妹妹病史与先证者类似。该家系中的3例患者因四肢功能表现异常入院治疗,疑似肌张力障碍,但是具体原因仍需进一步检查和确诊。
1.2方法
1.2.1外周血全基因组DNA的提取 根据知情同意原则,获取该核心家系中所有成员的抗凝外周静脉血5 mL,采用AxyPrepTMBlood genomic DNA Miniprep Kit试剂盒(购自江苏康宁生命科学有限公司)提取基因组DNA。
1.2.2WES检测 采用IIlumina NovaSeq高通量双端(Pair-end 150bp)测序平台(购自北京贝瑞和康生物技术有限公司)对先证者基因组DNA全外显子组进行WES检测,其简要流程如下:将1 μg基因组DNA随机打断成150~200 bp的片段,然后通过PCR扩增进行标本标记和富集DNA,将构建好的文库与已标记的RNA探针进行液相杂交,利用标记磁珠捕获目标区域,构建小片段测序文库。测序检测完成后,经碱基识别转化为原始序列数据,通过变异位点检索系统和变异位点注释系统对测序数据进行数据产量统计及单核苷酸多态性、插入和缺失检测及注释。以2015年美国医学遗传学与基因组学学会(ACMG)发布的遗传变异分类标准与指南及2016年发布的二级发现报告指南SFv2.0为指导[3],参考单核苷酸多态性数据库和人类基因突变数据库,筛选致病突变位点。根据SIFT、Polyphen-2、Mutation Taster软件对其突变位点进行评分并预测其致病性。SIFT分值为该变异对蛋白质的影响,分值越小越有害;Polyphen-2分值为Human Div数据库预测该变异对蛋白序列影响的分值,适用复杂表型中罕见等位基因位点突变的诊断,分值越大越有害,表明该单核苷酸多态性导致蛋白结构或功能改变的可能性越大;Mutation Taster分值为Mutation Taster软件的预测结果,分值越大结果越可靠。
1.2.3线粒体融合蛋白2(MFN2)位点Sanger测序验证 用PCR-Sanger测序对该核心家系4例患者进行突变位点的检测。在美国国立生物技术信息中心上在线合成1对包含MFN2基因(NM_014874.3)c.1090C>T p.R364W突变位点的特异性扩增引物,其中,正向引物序列为5′-TCC CTG GCA GTG AAA ACC AG-3′,反向引物序列为5′-CAG GGA AAG GGC TCT GGA TG-3′。40 μL的反应体系为40 ng基因组DNA约10 μL,10 μmol正反向引物各1 μL,2×PCR Master Mix 20 μL,用去离子水补足至40 μL。PCR反应条件为94 ℃预变性2 min,94 ℃ 45 s;55 ℃ 30 s,72 ℃ 30 s,30个循环;72 ℃ 延伸5 min。PCR产物经凝胶鉴定后进行Sanger测序。采用Geneious 11.1.5生物信息学软件对测序结果进行序列比对和分析。
2 结 果
2.1临床表型 检测结果显示先证者四肢肌张力升高,双上肢肌力Ⅳ~Ⅴ级,双下肢肌力Ⅳ级,姿势异常:前臂内旋,马蹄内翻足,踝关节趾曲、翻转,足趾屈曲,步态蹒跚不稳,下肢无力,手指关节挛缩无法伸直。四肢多条受检神经运动、感觉传导功能异常,表现为:(1)双侧胫、腓总、正中、尺神经复合肌肉动作电位(CMAP)均未引出;双侧股、桡神经CMAP波幅降低;(2)各受检神经F波均未引出;(3)各受检神经感觉神经动作电位均未引出。神经传导检测结果提示有四肢广泛性周围神经病变,传导阻滞,轴索病变为主可能,远端重于近端。
2.2WES结果 本研究利用WES技术对先证者进行检测并分析其结果,根据ACMG指南,在人类外显子数据库东亚人群(ExAC_EAS)中未发现MFN2基因c.1090C>T突变;经计算机辅助药物设计和神经网络领域对抗性训练等对其进行保守性预测,结果显示该位点进化上保守,具有潜在的功能影响;该位点致病性预测皆显示有害。提示先证者MFN2基因c.1090C>T为1个疑似致病的突变位点。 见表1。

表1 MFN2基因c.1090C>T p.R364W突变位点致病性预测
2.3Sanger测序结果 PCR-Sanger测序结果显示先证者、母亲及妹妹均为MFN2基因单碱基杂合突变c.1090C>T p.R364W,其父亲在该位点为野生型,未见碱基突变,表明WES预测正确,见图1。
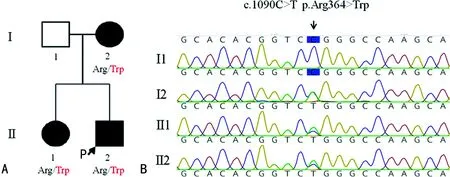
注:A为CMT患者家系图谱,一共4名成员,包括3例患者,Ⅰ1、Ⅰ2、Ⅱ1、Ⅱ2分别为父亲、母亲、先证者妹妹及先证者(箭头指示为先证者);Arg/Trp为精氨酸到色氨酸的氨基酸改变;B为MFN2基因PCR-Sanger测序结果。
3 讨 论
CMT是一类周围神经遗传性感觉损伤和运动神经损伤的疾病。周围神经可以将大脑和脊髓连接到肌肉和末端感觉细胞,这些细胞可以感知触摸、疼痛、热量和声音等。周围神经轴突的异常会引起肌张力异常,腓骨进行性肌肉萎缩足部畸形,远端无力,感觉丧失和视神经萎缩等[4]。随着疾病的发展,患者小腿的肌肉通常会萎缩,到晚期可能需要使用轮椅。CMT患者通常对足和小腿的触摸、疼痛及热量的敏感性较低,但偶尔会感到疼痛或灼痛感。在极少数情况下,受影响的患者会失去视力或逐渐丧失听力,甚至导致患者耳聋。尽管在大多数患者中,CMT不会影响预期寿命,较多患者仅产生中等程度的身体残疾,症状轻微,甚至可能不会出现明显的临床表型。但是,在极少数情况下,CMT也有可能会危及患者生命[5]。
根据遗传方式,CMT可表现为常染色体显性和隐性,以及X-连锁显性和隐性遗传,其中以常染色体显性遗传多见,此外还有少数纯合或复合杂合突变。根据神经病理学和电生理学标准,CMT被分为2个类型:脱髓鞘型(CMT1)和轴突型(CMT2),CMT2又包括CMT2A 、CMT2B和CMT2D等[6]。CMT2A(OMIM:608507)中最常见是MFN2基因突变,约占所有基因突变的20%。MFN2是CMT2主要的致病基因,突变频率为8%~30%。MFN2是编码线粒体GTP酶的核基因,与线粒体融合蛋白1、神经节苷脂诱导分化相关蛋白1和视神经萎缩1相互作用,对线粒体的结构完整性、形态和运输至关重要[7]。MFN2通过影响氧化磷酸化而参与能量代谢[8]。MFN2与Ca2+的吸收调节有关,促进内质网-线粒体的结合[9]。已有研究报道了罕见的MFN2复合杂合或纯合突变病例[10]。这有助于扩大与MFN2相关的神经疾病的遗传特征范围。在临床上,与MFN2相关的CMT2A发病特征可以从轻度的迟发性神经疾病到严重的早发性神经疾病[11],其表型通常比单纯的周围神经疾病复杂,有听力损失、视神经萎缩或锥体束征等其他特征[12]。有研究报道了15例CMT2A患者,他们均是MFN2突变的复合杂合子或纯合子携带者,具有常染色体隐性遗传或半显性遗传,这些患者通常表现出严重的早期发作性神经疾病,也有较轻的儿童期发病病例,有严重表型的患者通常会在30岁左右因下肢瘫痪需要乘坐轮椅,并合并其他发病特征[13-15]。已有研究表明,MFN2基因突变位点c.1090C>T的主要临床症状是视神经萎缩等,该位点位于MFN2蛋白高度保守的R3区,是1个突变热点[16]。
在本研究的核心家系中,先证者、母亲及妹妹均为MFN2基因c.1090C>T单碱基杂合突变,其父亲在该位点为野生型,未见碱基突变。先证者主要显示出轻度的营养不良和下肢无力,发病时间为3岁左右;先证者母亲8~9岁逐渐出现姿势异常及肌肉萎缩,后出现瘫痪,现仅能独坐,不能站立和行走,四肢纤细、消瘦,肌容积明显减少;妹妹病史与先证者类似;先证者父亲表型未见异常。先证者及其妹妹都表现出较早、较轻的儿童期发病,目前已经采用手术矫正的方法逆转其手足畸形,并配合其他方法尽量维持其肌肉的自主能力[17]。其母亲因发病时间较长且前期未做相关检查和治疗,目前已出现下肢瘫痪,无法行进一步干预。由于目前无法采集到除该核心家系以外的先证者其他家族成员的基因,所以仅能推断先证者及其妹妹的杂合突变来源于其母亲,但是无法推断其母亲突变基因的来源,需要扩大其家庭的临床、神经生理和分子研究,从而确定致病作用和遗传模式的可能性。因此,在向患者及其家属披露基因检测结果和遗传咨询之前,需要仔细收集家族史并对健康的亲属进行分析。此外,由于外显率不完全,复发风险可能大不相同,必须将有关基因检测的不确定性充分告知患者,以便患者可以更好地了解自己的病情。
综上所述,本研究基于患者的临床表型,以WES和PCR-Sanger测序方法,在1个核心家系中检测并鉴定了3例携带MFN2基因单碱基杂合突变的患者。之前的报道显示此突变主要是以视神经萎缩为主要临床症状,与本研究中报道的四肢发病特征不同,也证实了CMT具有明显的临床异质性,常难以明确认知和诊断,需要与其他疾病正确进行区分。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
现代园艺(2017年21期)2018-01-03
广东海洋大学学报(2015年4期)2016-01-13
中国康复理论与实践(2015年10期)2015-12-24
听力学及言语疾病杂志(2015年5期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
重庆医学(2015年12期)2015-03-05
