
纤维素类生物质制备乙酰丙酸酯和γ-戊内酯的研究进展
2021-04-15 09:14曾宪海
厦门大学学报(自然科学版) 2021年2期
唐 兴,孙 勇,曾宪海,林 鹿
(厦门大学能源学院,福建省生物质清洁高值化技术工程研究中心,福建 厦门 361102)
目前人类社会的发展严重依赖于煤、石油和天然气等非可再生化石资源.化石资源供应了80%以上的全球能源消耗和90%的有机化学品消耗.随着全球人口的持续增长及人们对提高自身生活水平的不断追求,预计人类每年对化学品、材料及能源的需求增速将达到7%左右[1].BP公司发布的《2019年世界能源统计》中的数据显示:截至2018年底,世界石油和天然气探明可采剩余储量分别为17 300亿桶和1 969 000亿m3,其储采比(储量/产量)分别为50.0和50.9年;煤炭探明可采剩余储量为10 547.82亿t,储采比约为132年[2].从长远来看,随着能源资源供求关系的紧张,化石资源的价格将会不断攀升.此外,化石资源在开采、加工和使用过程中往往对生态环境造成较严重的破坏和污染,最终将威胁到人类自身的生存和发展.人类必须实现社会经济和生态环境可持续性发展的转型,开发利用可持续供应的清洁能源资源,包括生物质能、光伏太阳能、核能及风能等.其中,生物质是唯一可以实现化石资源全替代的可再生资源,可以供应包括食物、电、热、化学品、材料及液体燃料等人类必需的几乎所有能量形式[3-4].全球每年的生物质量高达1 700亿t,折合能量相当于每年世界石油产量的15~20倍,然而其中仅约3%的生物质资源被人类利用(包括食物和非食用途径)[5].因此,开发利用生物质能对提高能源供应安全、改善生态环境及人类社会的可持续发展等方面都具有深远的意义.
含氧生物燃料是生物质利用的重要方向,这些含氧燃料可以作为常规汽柴油的添加组分以改善燃料燃烧性能,同时可以替代一部分化石燃料的使用.近年来,生物质基乙酰丙酸酯和γ-戊内酯作为酯类生物燃料受到广泛关注.如表1所示:乙酰丙酸甲酯和γ-戊内酯的沸点分别为196 ℃和207~208 ℃,都处于或接近常规汽柴油的沸程范围;此外,二者都具有较高的辛烷值和与汽柴油接近的能量密度.乙酰丙酸甲酯和γ-戊内酯的沸点和能量密度远高于乙醇,且具有与乙醇接近的辛烷值.值得注意的是,微生物利用一分子葡萄糖发酵代谢生产乙醇的过程会产生两分子CO2,这造成了较大的碳原子损失;而一分子葡萄糖化学转化制备乙酰丙酸甲酯和γ-戊内酯只损失一分子碳.因此,纤维素类生物质转化制备乙酰丙酸甲酯和γ-戊内酯是更具原子经济性的生物燃料生产途径.
乙酰丙酸酯的性质与生物柴油相似,可以与石化柴油和生物柴油等混合作燃料,添加后能有效改善燃烧清洁度,且具备优良的润滑能力、闪点稳定性和低温流动性[6].Horvth等[7]认为γ-戊内酯是比乙醇更好的燃料添加剂,因为γ-戊内酯具有更低的饱和蒸汽压和更高的能量密度;且不同于乙醇的是,γ-戊内酯与水不会形成共沸物,因此在水溶液中浓缩提纯γ-戊内酯可能比从发酵液中分离提纯乙醇更容易.Bruno等[8]系统地研究了γ-戊内酯与AI-91号夏季汽油组成的混合燃料的各项性能,发现γ-戊内酯的加入能够大大降低排放尾气中CO和烟尘的浓度.因此,乙酰丙酸酯和γ-戊内酯也是性能优异的生物燃料组分.

表1 乙酰丙酸甲酯和γ-戊内酯的理化性质Tab.1 Physicochemical properties of methyl levulinate and γ-valerolactone
2017年9月13日,国家发展改革委员会、国家能源局、财政部等15个部门联合印发《关于扩大生物燃料乙醇生产和推广使用车用乙醇汽油的实施方案》,明确提出:2020年,车用乙醇汽油要在全国范围内基本实现全覆盖.然而,由于燃料乙醇的市场供应缺口高达每年千万吨,该目标难以达成.这是因为:1) 粮食乙醇的大规模生产容易陷入与民争粮的困境,在遭遇重大自然灾害对国内和国际粮食供应造成压力的情况下尤为如此;2) 纤维素乙醇成本依然较高,企业行业推广工业规模纤维素乙醇生产的动力不足.因此,开发经济高效的纤维素类生物质转化合成乙酰丙酸酯和γ-戊内酯等新型生物含氧燃料的技术途径,有助于拓展含氧生物燃料种类的多样性,并在此基础上获得性能更优的生物燃料.
此外,乙酰丙酸酯和γ-戊内酯作为一类多功能的生物质基平台分子,转化制备其他高附加值化学品、材料和烃类燃料的应用同样受到大量关注和研究[9].以γ-戊内酯为例,其经开环加氢后可得到戊酸和甲基四氢呋喃[10-11];通过连续的缩聚以及加氢脱氧,可以制备某些C8~C18的分支或直链烃类燃料[12];还可以转化合成α-亚甲基-γ-戊内酯和ε-己内酰胺等多种聚合单体[13-14].本文详细总结了近年来纤维素类生物质在多种酸催化体系中制备乙酰丙酸酯的研究进展,并从氢源的角度探讨其对乙酰丙酸(酯)加氢合成γ-戊内酯的影响.
1 纤维素类生物质转化为乙酰丙酸酯和γ-戊内酯的反应途径
纤维素类生物质中的纤维素和半纤维素组分都可用于制备乙酰丙酸酯,乙酰丙酸酯经催化加氢后可以合成γ-戊内酯[15].如图1所示:半纤维素中的木糖部分在酸催化下脱水可以得到糠醛,糠醛经过催化加氢可以制备糠醇[16],最终糠醇在水相或醇溶液中经酸催化水解或醇解可分别得到乙酰丙酸或乙酰丙酸酯[17-18];而纤维素、葡萄糖或果糖在水相中经酸催化降解可以制备5-羟甲基糠醛[19],5-羟甲基糠醛进一步水合可以得到乙酰丙酸[20];此外,纤维素、葡萄糖或果糖在醇溶液中经酸催化醇解也可以直接制备乙酰丙酸酯[21-22].通过上述途径制备的乙酰丙酸及其酯类可以通过催化加氢合成γ-戊内酯.
2 纤维素类生物质直接醇解合成乙酰丙酸酯
纤维素作为自然界中最丰富的生物大分子,是用于制备乙酰丙酸酯廉价易得的原料;但纤维素分子质量较大,通常不溶于水及一般有机溶剂,造成其化学反应的效率和选择性较差.尤其是木质纤维生物质原料,其纤维素与半纤维素、木素之间相互交织形成致密的网状结构,并且相互间有氢键、范德华力和化学共价键的连接,利用其直接醇解合成乙酰丙酸酯,首先需要使纤维素分离暴露出来,才能保证下一步的醇解反应高效进行.目前已有较多关于纤维素类生物质直接醇解合成乙酰丙酸酯的研究报道,开发的催化体系主要有无机酸催化、固体酸催化和混合酸催化体系.

图1 从纤维素和半纤维素制备乙酰丙酸酯和γ-戊内酯的反应路径Fig.1 Reaction pathways for the production of alkyl levulinates and γ-valerolactone from cellulose ann hemicellulose
2.1 无机酸催化
由于无机酸较易与纤维素固体发生作用,且成本低、容易获取,所以得到较广泛的应用和研究.Garves[23]较系统地研究了在180~200 ℃高温下酸催化降解纤维素转化合成乙酰丙酸酯的反应情况,发现稀硫酸(质量分数<5%)对纤维素醇解具有较好的催化活性,在不同醇体系中产物乙酰丙酸酯的理论得率均可达37%以上.稀硫酸还可以高效催化刨花板、麦秆等生物质原料醇解制备乙酰丙酸,乙酰丙酸酯的得率可达50%左右[24-25].
降低硫酸浓度有助于降低对反应设备的要求,但通常反应需要在更严苛的条件下进行.例如,采用超低硫酸体系(质量分数0.1%)在210 ℃下反应2 h,纤维素转化成乙酰丙酸甲酯的得率也可达50%[26].相较之下,在较高浓度的硫酸体系中可以催化纤维素醇解反应在更温和的条件下进行.例如,在20%(质量分数)的硫酸丁醇体系中,于130 ℃下反应20 h,纤维素制备乙酰丙酸丁酯的得率可达62%;进一步提高硫酸质量分数至30%,在5 h内乙酰丙酸丁酯的得率即可达60%[27].
2.2 固体酸催化
纤维素类生物质原料不溶于醇类介质,由于固固传质阻力,固体酸在醇溶剂中难以与纤维素相互接触,限制了固体酸的应用.通常纤维素需要先经过前处理或在更苛刻的反应条件下才能发挥固体酸的催化活性.例如,Rataboul等[28]在超临界条件下(300 ℃/10 MPa)研究了多种固体酸在甲醇中直接催化纤维素制备乙酰丙酸甲酯的效果,结果显示固体酸CsxH3-xPW12O40或SO42-/ZrO2催化1 min,乙酰丙酸甲酯得率仅为16%~20%.而在常规溶剂热条件下(180~200 ℃,4 h),硫酸化蒙脱土催化纤维素醇解转化为乙酰丙酸甲酯的得率也只有24%~27%[29].此外,近年来研究人员开发了一系列新型的固体酸催化剂,包括酸性分子筛HUSY、磺酸功能化的ZrO2及磺化的碳基催化剂等,这些催化剂在催化碳水化合物醇解制备乙酰丙酸酯方面都具有较好的催化活性[30-33].但需要注意的是,固体酸催化不溶性纤维素醇解的反应效率较低,通过延长反应时间可以在一定程度上提高乙酰丙酸酯得率.例如,在180 ℃反应24 h的条件下,铌基磷酸盐在甲醇中催化纤维素醇解制备乙酰丙酸甲酯的得率可达56%[34].
2.3 混合酸催化
最近,研究发现Brønsted酸结合Lewis酸的混合酸体系可高效地催化纤维素类生物质直接醇解制备乙酰丙酸酯[35].在Lewis酸三氟甲磺酸盐和Brønsted有机酸混合协同催化下,纤维素醇解转化为乙酰丙酸酯的选择性和得率大大提高[36-37].例如,在三氟甲磺酸铟和对甲苯磺酸的协同催化下,纤维素醇解转化为乙酰丙酸甲酯的得率可达70%(180 ℃,5 h);如改用2-萘磺酸代替对甲苯磺酸则乙酰丙酸甲酯的得率可进一步提高至75%[36].进一步研究表明,Brønsted有机酸能催化纤维素中β-1,4-糖苷键及氢键断裂进而得到葡萄糖,而Lewis酸三氟甲磺酸盐则可有效催化葡萄糖异构化成果糖,继而进一步反应生成乙酰丙酸酯[36-37].此外,这种混合酸催化体系也可用于松木、桉木及甘蔗渣等木质纤维生物质原料的转化,其乙酰丙酸甲酯得率均可达70%左右[38].然而,上述均相的混合酸催化剂难以回收利用,且价格昂贵的三氟甲磺酸盐会影响该技术的实际应用.最近研究[39-40]发现杂多酸和Sn-beta分别可以作为Brønsted酸和Lewis酸催化纤维素或葡萄糖醇解制备乙酰丙酸酯,在160~180 ℃下乙酰丙酸酯的得率超过60%,且Lewis酸催化剂方便回收利用.
以上3种催化体系对纤维素类生物质直接醇解合成乙酰丙酸酯的代表性结果如表2所示.

表2 纤维素类生物质制备乙酰丙酸酯的代表性结果Tab.2 Representative results for the production of alkyl levulinates from cellulosic biomass
3 乙酰丙酸(酯)催化加氢制备γ-戊内酯
如图1所示,纤维素和半纤维素都可以转化为乙酰丙酸(酯),其经过催化加氢后可以合成γ-戊内酯.近年来,已有大量研究关注于催化乙酰丙酸(酯)选择性加氢合成γ-戊内酯.依据氢源的差异可以将这些催化反应体系分为3类:H2作为外部氢源、甲酸作为原位氢源和醇类作为原位氢源的体系.
3.1 H2作为外部氢源
图2描述了分子H2催化体系中乙酰丙酸选择性还原制备γ-戊内酯的两种可能机理.一般认为在液相加氢体系中,乙酰丙酸分子中的4位羰基首先被还原成羟基得到4-羟基戊酸,4-羟基戊酸对热不稳定,很容易继续环化脱去一分子水形成更稳定的γ-戊内酯[42];而在气相加氢体系中,较高的气化温度会导致乙酰丙酸发生烯醇化并脱水环化形成α-当归内酯,α-当归内酯进一步加氢还原可以生成γ-戊内酯[43].

图2 乙酰丙酸选择性加氢还原合成γ-戊内酯的可能反应机理Fig.2 The probable mechanism for the production of γ-valerolactone by selective hydrogenation reduction of levulinic acid
催化乙酰丙酸(酯)加氢还原合成γ-戊内酯的催化剂可以分为均相催化剂和非均相催化剂.应用均相催化剂合成γ-戊内酯的研究可以追溯到1991年,Braca等[44]利用Ru(CO)4I2和共催化剂NaI在水相中催化乙酰丙酸加氢还原,γ-戊内酯的最高得率能达到87%(150 ℃,8 h).进一步研究发现,相对于其他贵金属催化剂,Ru基催化剂对催化乙酰丙酸还原合成γ-戊内酯具有非常高的活性和选择性,在室温甚至无溶剂的反应条件下都具有很好的催化效果[45-46].但均相的Ru基催化剂通常需要在配体或助催化剂存在时才能发挥稳定的催化性能[47-48],并且贵金属均相催化剂的回收再利用也比较困难[49].基于此,van Slagmaat等[50]开发了一种可回收的Ru基Shvo催化剂,不仅可以高效催化乙酰丙酸(酯)无溶剂加氢合成γ-戊内酯,而且当以乙酰丙酸乙酯为底物时能实现催化剂的回收利用.此外,Ir基的配位催化剂对乙酰丙酸选择性还原也具有非常高的催化活性,在水相或乙醇中γ-戊内酯的得率均可达95%以上[51-52].最近,Yi等[53]研究发现非贵金属的Fe基配位催化剂也能高效催化乙酰丙酸(酯)加氢合成γ-戊内酯,通过调控配体后,Fe基配位催化剂在甲醇中催化乙酰丙酸加氢合成γ-戊内酯的转化频率(TOF)和转化数(TON)分别高达1 917 h-1和23 000(100 ℃,12 h).但需要注意的是,Fe基配位催化同样需要较为复杂的配体和碱促进剂,催化体系的回收利用也存在较大难度.
从经济性角度考虑,特别是对于贵金属基催化剂而言,非均相催化剂能够比较容易实现回收再利用,节约生产成本.因此,目前选择性还原乙酰丙酸(酯)合成γ-戊内酯的研究主要关注于非均相的催化体系,如表3所示.早在1930年,Schuette等[71]就在较低H2压力和长反应时间条件下以PtO2定量催化乙酰丙酸还原合成γ-戊内酯.与均相催化剂体系一样,负载型的Ru基催化剂(如Ru/C)比Pt/C等其他负载型贵金属催化剂对催化乙酰丙酸的选择性还原具有更高的活性,并且在Ru/C催化下γ-戊内酯的得率接近100%[67,72-73].除了活性金属的影响外,载体的性质对负载型催化剂的催化活性同样有重要的影响.Al-Shaal等[68]在各种醇和水及其混合体系中检验了不同载体的Ru基催化剂加氢还原乙酰丙酸的能力,研究发现在相同的反应条件下Ru/C催化合成γ-戊内酯的得率能达到97.5%,而Ru/Al2O3或Ru/SiO2催化下的γ-戊内酯得率分别只有1.7%和6.3%.这是由于相对于其他载体,Ru纳米颗粒在活性炭上具有更高的分散度[63],高分散性的纳米颗粒提供了更多的催化活性中心,所以具有更高的催化效率.
为了保证乙酰丙酸能够高效地转化生成γ-戊内酯,Ru基催化剂催化反应的初始氢压通常需要在2 MPa 以上,反应温度则需要150 ℃左右.降低反应温度或初始氢压能够极大地降低对反应设备的要求和能耗,进而提高加氢工艺的经济性.Raspolli Galletti等[64]发现Ru/C在固体酸协助下可以在较低的反应温度下催化乙酰丙酸选择性还原制备γ-戊内酯;当Amberlyst A70作为共催化剂时,在70 ℃、0.5 MPa H2条件下反应3 h,γ-戊内酯的得率仍可以高达97%以上,而在相同反应条件下无固体酸共催化剂存在时,γ-戊内酯的得率不到15%.事实上乙酰丙酸分子中4位羰基的加氢还原在较低温度下(<100 ℃)就能完成,而加氢中间产物4-羟基戊酸需要在较高的温度下(约150 ℃)才能继续环化脱水形成γ-戊内酯.4-羟基戊酸的环化反应本质上是一个分子内的酯化反应,因而酸性催化剂如固体酸能明显促进环化反应在较低的温度下进行.基于这一原理,很多富含酸性催化位点的载体被用于制备负载型Ru基催化剂,如磺化阳离子交换树脂、羟基磷灰石、磺化聚醚砜、Nb2O5及分子筛(如H-ZSM5和H-beta等)[55,65,74-78].这些酸性载体负载的Ru基催化剂能将乙酰丙酸加氢环化合成γ-戊内酯的反应温度和初始氢压分别控制在100 ℃和1 MPa以内.而当反应温度和初始氢压过高时,酸性的催化剂载体则能进一步催化γ-戊内酯开环加氢形成戊酸、戊烯酸、1,4-戊二醇和甲基四氢呋喃等副产物[69].

表3 外加H2下乙酰丙酸(酯)加氢合成γ-戊内酯的代表性非均相体系Tab.3 Representative heterogeneous systems for the production of γ-valerolactone from alkyl levulinates hydrogenation using external H2
然而,Ru等负载型贵金属催化剂在使用过程中难以避免其活性金属的流失.用便宜的非贵金属催化剂替代Ru基催化剂同样可以提高加氢反应的经济性.例如,研究发现Cu[57-58,62]、Mo2C[79]、Ni[80]和Co[81]等非贵金属催化剂对乙酰丙酸选择性加氢还原合成γ-戊内酯均具有较高的催化活性,其催化下的γ-戊内酯最高得率可以达到95%以上;但是这些非贵金属催化剂通常需要在较高的反应温度(约200 ℃)和初始氢压(约4 MPa)的条件下才能有效地催化乙酰丙酸还原合成γ-戊内酯,这在一定程度上抵消了催化剂成本相对于贵金属的经济优势.因此,设计合成在温和条件下具备高效催化乙酰丙酸(酯)加氢合成γ-戊内酯的非贵金属催化剂,将是今后研究的重要方向之一.例如,Fang等[82]最近开发了一种以生物质如柚子皮作为还原碳源制备Ni/C催化剂的方法,通过该方法制备的Ni/C催化剂在水相中160 ℃下催化乙酰丙酸加氢合成γ-戊内酯的得率超过90%.
3.2 甲酸作为原位氢源
根据碳水化合物酸水解的反应机理,每产生1 mol 乙酰丙酸,会伴随产生等物质的量的甲酸[20,83].催化甲酸分解可以得到H2和CO2混合气体产物,因此可以应用甲酸作为原位氢源还原乙酰丙酸合成γ-戊内酯,这也是原子经济性原则的体现,代表性结果如表4所示.最近,Mehdi等[46]发现在pH=4的HCOONa水溶液中,配位催化剂[(η6-C6Me6)Ru(bpy)(H2O)][SO4]能够在无外部H2存在下催化乙酰丙酸还原合成γ-戊内酯;但是上述反应需要在惰性气氛中进行,γ-戊内酯的得率也只有25%,另外过度加氢产物如1,4-戊二醇等的得率也达到25%左右.在碱性促进剂(如KOH和NH3等)和配体(如PPh3等)可调的均相Ru基催化剂体系中,甲酸原位产氢还原乙酰丙酸合成γ-戊内酯的得率可以提高至90%以上[86],但是这种催化体系相对复杂,且催化剂难以回收利用.此外,研究发现Shvo催化剂对催化甲酸产氢和乙酰丙酸加氢也具有较高的活性[92],然而Shvo催化剂同样价格昂贵且不易回收.最近,Amenuvor等[93-94]提出了一种Ru基配位催化体系的回收方法:反应完成后先将反应混合物溶解于甲醇或乙醇中,然后在减压蒸馏条件下分离相对低沸点的醇、水、γ-戊内酯和碱促进剂,剩下的高沸点Ru基配位催化剂可重新用于下一次反应.该回收方法可以实现负载型催化剂重复利用3~7次,但催化剂与反应产物分离的能耗较高.
非均相催化剂如Ru/C、Pt/ZrO2、Ni-Au合金、Ni/Cu-SiO2和Ag-Ni-ZrO2也能有效地催化甲酸分解产氢,并同时还原乙酰丙酸合成γ-戊内酯[85,95-98],然而这些催化剂一般需要在甲酸过量的条件下才能催化产生足够多的H2用于乙酰丙酸的全部转化.值得注意的是,Du等[87]制备的Au/ZrO2在150 ℃下就能高效地催化纤维素水解产生的甲酸原位分解产氢并同时还原乙酰丙酸,且上述反应体系不需要引入过量甲酸或外部H2.最近,Cu/ZrO2同样被发现具有催化甲酸原位分解产氢和还原乙酰丙酸的活性,但是其催化效率远远低于Au/ZrO2且需要在较高的反应温度(200 ℃)下才能完全转化乙酰丙酸[89].此外,半纤维素及其衍生化合物也可以甲酸作为原位氢源合成γ-戊内酯.例如,Wang等[99]设计合成的壳聚糖螯合的Ru基配位催化剂在温和条件下可以催化糠醛和甲酸一锅转化合成γ-戊内酯(得率可达79%),该催化剂与酸性分子筛如ZSM-5组合的催化体系可以进一步将半纤维素或木糖转化合成γ-戊内酯.
在上述研究中,催化剂中的活性金属扮演着双重催化作用,即同时催化甲酸分解产氢和乙酰丙酸加氢还原.以甲酸作为氢源其本质上还是以分子H2加氢还原乙酰丙酸合成γ-戊内酯,只是这里的H2来自于制备乙酰丙酸过程中所产生的副产物甲酸.

表4 甲酸作为原位氢源催化乙酰丙酸(酯)合成γ-戊内酯的代表性催化体系Tab.4 Representative catalytic systems for the production of γ-valerolactone from alkyl levulinates using formic acid as the hydrogen source
3.3 醇类作为原位氢源
乙酰丙酸(酯)加氢还原合成γ-戊内酯的反应本质上是一个羰基选择性还原的过程(图2).除分子H2外,脂肪醇类也可以作为氢供体,并通过Meerwein-Ponndorf-Verley(MPV)反应催化羰基化合物转移加氢合成相应的醇类.MPV转移加氢反应对羰基具有专一选择性,因此MPV反应在不饱和醛酮的选择性还原反应中具有广泛的应用[100-101].代表性的乙酰丙酸(酯)经MPV还原合成γ-戊内酯的结果如表5所示.最近,Chia等[102]以金属氧化物催化醇类(乙醇、2-丁醇、异丙醇等)氢转移还原乙酰丙酸酯,γ-戊内酯的得率可达80%以上.在众多的金属氧化物中,以ZrO2的催化活性最佳.然而,以乙酰丙酸作为反应底物时,即使在220 ℃下经过长达16 h的反应后γ-戊内酯得率也只有71%,这主要是由于ZrO2的催化活性与催化剂表面酸碱活性位点密切相关,而乙酰丙酸属于酸性较强的有机酸,所以可能与催化剂表面的碱性位点发生相互作用导致催化剂部分失活[103-104].Zr-beta分子筛也能有效地催化乙酰丙酸酯经MPV转移加氢反应合成γ-戊内酯[105-106],但是Zr-beta分子筛的制备工艺要比金属氧化物复杂.ZrO2也可以负载到SBA-15等其他分子筛上,得到具有较高转移加氢活性的负载型催化剂[108].值得注意的是,Tang等[109]开发的原位催化剂体系能够高效地催化乙酰丙酸在醇体系中转移加氢合成γ-戊内酯.在这种催化剂体系中,催化剂前体ZrOCl2·8H2O在乙酰丙酸的醇溶液中受热自发分解为HCl和ZrO(OH)2,并可分别有效地催化乙酰丙酸的酯化和后续酯化产物的转移加氢.这种原位催化剂体系避免了繁琐的催化剂制备过程,特别是原位形成的催化剂比传统沉淀法制备的氢氧化物具有更高的比表面积,并且对腐殖质也具有较好的耐受性.
Zr4+是催化MPV还原的活性Lewis酸中心,因此获得高度分散的Zr4+中心有利于提高催化剂的催化活性.Song等[110]利用ZrCl4与含有多个磷酸基团的植酸反应,合成得到具有介孔结构的配位聚合物材料Zr-PhyA,其中配位聚合物金属节点中心Zr4+具有高度的分散性,而与Zr4+中心配位的磷酸基团可以诱导Zr4+中心周围电子向磷酸基团偏移,进一步增加了Zr4+中心的Lewis酸性,可使Zr-PhyA在温和条件下催化乙酰丙酸乙酯转移加氢合成γ-戊内酯的得率达到96.7%(150 ℃,6 h).通过调整这类Zr基配位聚合物配体的结构可以比较灵活地调控催化剂的活性.例如,以三聚氰酸作为配体合成的Zr-CA甚至可以在更温和的反应温度(130 ℃)下高效催化乙酰丙酸转移加氢合成γ-戊内酯[111].也可以调整这类配位聚合物催化剂的Lewis酸中心金属离子.例如,最近Li等[112]和Zhou等[113]以Hf作为Lewis酸金属中心合成的多孔性配位聚合物催化剂,同样具备高效催化乙酰丙酸酯转移加氢的性能.此外,通过螯合的方式也可以制备高度分散的Zr4+Lewis酸中心.例如,Wang等[114]以结构中富含—NH2和—OH的壳聚糖作为螯合Zr4+的模板载体,得到的螯合催化剂同样具有高度分散的强Zr4+Lewis酸中心,研究还发现螯合基团—NH2可以与Zr4+形成Lewis酸碱对,进一步促进转移加氢反应的进行.
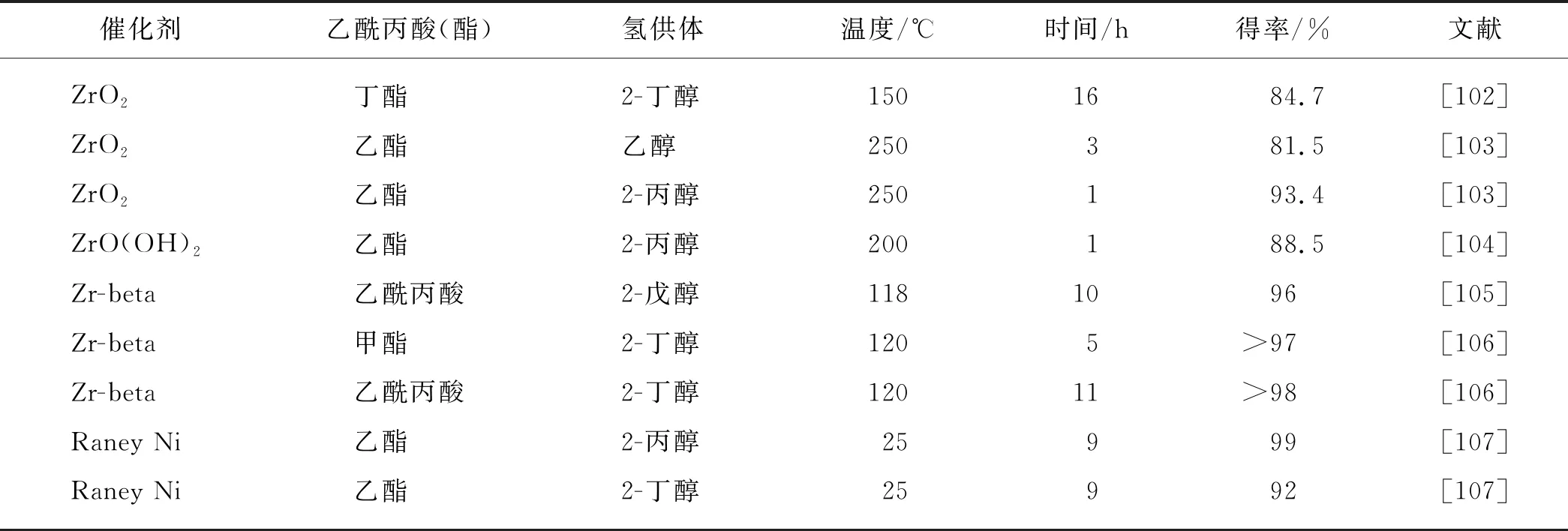
表5 乙酰丙酸(酯)经MPV还原合成γ-戊内酯的代表性催化体系Tab.5 Representative catalytic systems for the production of γ-valerolactone from alkyl levulinates via MPV reduction
此外,Yang等[107]制备的Raney Ni在室温条件下就能催化乙酰丙酸乙酯在异丙醇中转移加氢,γ-戊内酯的最高得率接近100%.但是Raney Ni催化氢转移机理不同于MPV还原,而更类似于催化H2加氢的机理.这种自制的Raney Ni在室温条件下的优异催化性能主要得益于其制备过程中残留的酸性组分γ-Al2O3,因为酸性组分能够极大地促进加氢中间产物的环化反应在低温下进行[115].Pd、Ru等贵金属基催化剂也能催化乙酰丙酸酯在醇溶液中转移加氢合成γ-戊内酯[116-118],但从催化剂成本方面考虑,贵金属催化剂不是最理想的选择.上述转移加氢途径通常只能有效地利用两个C以上的脂肪醇作为氢供体,而甲醇在MPV转移加氢反应中属于非常惰性的氢供体,在甲醇中乙酰丙酸酯通过MPV还原合成γ-戊内酯的得率一般都在10%以下.然而,Tang等[119]发现纳米Cu催化剂能够同时有效地催化甲醇重整制氢和乙酰丙酸甲酯还原加氢合成γ-戊内酯,并且在腐殖质存在下纳米Cu催化剂也能表现出比较稳定的催化性能.因此,通过纤维素甲醇醇解制备的乙酰丙酸甲酯粗产品的加氢还原,可以直接以溶剂甲醇作为原位氢源,从而省去了乙酰丙酸甲酯的分离提纯过程,极大地简化了生产工艺.
近年来越来越多的研究关注于利用脂肪醇类作为原位氢源合成γ-戊内酯.相对于传统外部H2的加氢途径,醇作为原位氢源的工艺消除了引入外部H2的单元操作和贵金属催化剂的使用,分别代之以更便于管理的醇类和便宜的过渡金属催化剂如Cu、Zr等.MPV转移加氢反应还可以消除原来分子H2与加氢底物和固体催化剂之间的气液、气固传质阻力,且不需要额外的输氢设备,有助于简化整体加氢工艺,提高其经济性.
4 结论与展望
乙酰丙酸酯和γ-戊内酯作为新型的含氧生物燃料,其在未来生物燃料领域中的应用潜力在很大程度上取决于其从生物质制备的成本.因此,需要开发高效经济的纤维素类生物质转化制备乙酰丙酸酯和γ-戊内酯的催化工艺技术,并在中试以上规模实验中验证技术的可行性和经济性,为未来大规模地应用乙酰丙酸酯和γ-戊内酯含氧生物燃料奠定基础.
基于绿色化学的理念,设计合成高性能的固体酸催化剂用于催化纤维素类生物质醇解制备乙酰丙酸酯应是未来研究的方向.由于纤维素类生物质醇解反应过程中不可避免地会产生分子量较大的腐殖质等副产物,所以这类固体酸催化剂对腐殖质的耐受性及其再生过程中的热稳定性对于提高催化剂性能和降低催化工艺成本尤为重要.此外,直接在纤维素类生物质水(醇)解反应液中催化乙酰丙酸(酯)加氢是一种更经济的γ-戊内酯合成途径,这样可以避免耗能的乙酰丙酸(酯)分离提纯.但需要注意的是,上述工艺的实现依赖于对腐殖质和甲酸等副产物具有良好耐受性的高效加氢催化剂.从氢源角度考虑,以副产物甲酸或溶剂醇作为原位氢源催化还原乙酰丙酸(酯)将进一步提高γ-戊内酯合成工艺的经济性.对于以分离纯化后的乙酰丙酸(酯)为原料合成γ-戊内酯,采用无溶剂催化剂体系有利于后续产物的分离提纯并消除溶剂的使用和回收.需要特别指出的是,设计合成对乙酰丙酸具有良好耐受性的高效加氢催化剂是乙酰丙酸高效无溶剂加氢的关键.
未来还应通过台架试验等深入研究乙酰丙酸酯和γ-戊内酯作为生物含氧燃料的综合燃烧性能,通过全生命周期评估等手段全方位地考察乙酰丙酸酯和γ-戊内酯作为生物含氧燃料的碳减排意义,逐步建立乙酰丙酸酯和γ-戊内酯掺混常规汽柴油的标准体系,并需要联合行业内相关企业争取乙酰丙酸酯和γ-戊内酯作为生物含氧燃料的市场准入,逐步在市场层面推广应用酯类生物燃料.
猜你喜欢
分子催化(2022年1期)2022-11-02
核化学与放射化学(2022年2期)2022-04-28
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
现代食品(2016年24期)2016-04-28
中国卫生标准管理(2015年5期)2016-01-14
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
云南中医学院学报(2015年2期)2015-07-31
医学研究杂志(2015年5期)2015-06-10
质谱学报(2015年5期)2015-03-01
