
碳自掺杂石墨相氮化碳纳米片的制备及其在可见光照射下的光解水产氢性能
2021-04-10 14:19上官莉谭宇烨孙建华刘光祥
无机化学学报 2021年4期
陆 杨 上官莉 张 慧 王 岩 谭宇烨 孙建华*, 刘光祥
(1江苏理工学院化学与环境工程学院,先进能源功能材料研究所,常州 213001)
(2南京晓庄学院环境科学学院,新型功能材料南京市重点实验室,南京 211171)
0 引 言
当前,在不断恶化的环境问题和全球能源危机的推动下,寻求清洁、环境友好的新型能源一直是个热门话题。氢能因为具有热效率高、能量密度大和绿色环保的优点,被认为是代替传统能源最适合的能源载体之一[1-2]。1972年,日本东京大学Fujishima和Honda两位教授首次报告以TiO2为光催化剂,利用太阳能直接分解水制氢,从而开辟了利用太阳能光解水制氢的研究道路[3]。其中,光催化剂是光解水制氢技术的关键[1]。
石墨相氮化碳(g-C3N4)是一种聚合物半导体,结构中的C、N原子以sp2杂化排列[4],具有密度低、硬度高和化学稳定性好等特点。而且g-C3N4的禁带宽度为2.7 eV,其导带(CB)和价带(VB)位置分别在-1.3和1.4 V(vs RHE)(pH=7),具有较强的光催化分解水产氢能力,成为了光解水半导体的新宠和研究热点之一[5-7]。但是由于g-C3N4带隙相对较宽,仅限于吸收太阳光谱中小于460 nm的蓝紫光;此外,还存在电荷转移率低和光生电子-空穴复合率高等不足,使得其光催化分解水制氢的效率不太理想[8-9]。为此,调节g-C3N4的能带结构,拓展其对可见光的吸收范围,是提升其光解水效率的关键因素。围绕这一主题,众多的科学工作者开展了大量而系统的研究,并已取得不错的进展。例如,通过元素掺杂和分子共聚等[10-13]方法,能够实现对g-C3N4能带结构调控,拓展其对可见光的吸收范围,从而有效提高其可见光光催化分解水产氢性能。
但是杂原子的引入会增加光生载流子重组位点等缺陷,从而降低了光生载流子的分离效率,导致光解水性能下降。因此,设计仅含碳、氮2种元素的自掺杂g-C3N4的改性策略是一项很有意义的工作。Dong等[14]发现碳自掺杂可以通过形成离域大π键,增加对可见光吸收和导电性,从而增强光氧化和还原活性。同时自掺杂避免了易于成为光生载流子复合中心的杂原子和缺陷的引入,从而提高光生载流子的分离效率[15-16]。
因此,我们设计一种碳自掺杂氮化碳的制备方案,通过在氮化碳共轭体系中引入富碳嘧啶环,拓宽氮化碳对可见光的吸收范围,提升对可见光的吸收效率;同时,降低氮化碳的阻抗,加速氮化碳光生载流子迁移速率,提高其光生载流子的分离效率,从而有效提高氮化碳的可见光光催化分解水产氢效率。
1 实验部分
1.1 试 剂
尿素、2,4,6-三氨基嘧啶(TAPD)、氯铂酸(H2PtCl6·6H2O)、三乙醇胺和乙醇均购自国药集团化学试剂有限公司。所有试剂均为分析纯,使用前未作任何处理。实验用水均为超纯水。
1.2 光催化剂的制备
称取10 g尿素于研钵中,加入5 mL无水乙醇,加入 x mg(x=10、30、50、100)2,4,6-三氨基嘧啶(TAPD),研磨混匀,将得到的粉末转移至50 mL坩埚,随后置于100℃烘箱中烘干,最后转移至马弗炉中以17℃·min-1程序升温至550℃后恒温4 h,并以1℃·min-1程序降至室温后得到π共轭碳自掺杂g-C3N4纳米片,产物命名为CNNS-x(x=10、30、50、100)。同CNNS-x的制备条件,前驱体中不加TAPD,得到石墨相氮化碳g-C3N4纳米片,简记为CNNS。
1.3 光催化剂的表征
采用德国蔡司Sigma 500型高分辨率场发射电子显微镜(FESEM,工作电压为20 kV,工作距离为8 mm)和日本电子JEM-2100型透射电子显微镜(TEM)观察催化剂的形貌。采用美国康塔公司的QUDRASORB SI型全自动独立多站比表面积和孔隙度分析仪(BET)分析样品的比表面积,测试前在120℃下对样品先进行脱气处理,然后在液氮温度(77 K)下进行测试。采用荷兰帕纳科公司的PW3040/60型X射线粉末衍射仪(XRD,Cu Kα,U=4 kV,I=40 mA,λ=0.154 06 nm,2θ=10°~80°)对样品的结构进行分析。采用德国Elementar公司的Inductar EL cube型元素分析仪(EA)和美国Thermo Fisher Scientific公司的EscaLab 250型X射线电子能谱仪(XPS)分析催化剂所含元素的含量及价态。采用美国珀金埃尔默公司的Lambda 950型固体紫外可见漫反射光谱仪(UV-Vis DRS)表征样品的光吸收性能。采用美国HORIBA的Fluoro Max-4型荧光光谱分析仪(PL)分析样品的荧光发光性能。
1.4 电化学和光电化学测试
用上海辰华CHI 760E型工作站进行光催化剂光电性能和电化学测试。测试采用三电极体系:Ag/AgCl电极为参比电极,Pt电极为对电极,制备的样品为工作电极。工作电极制备方法:称取5 mg样品分散在0.5 mL质量分数0.02%的Nafion乙醇溶液中,超声1 h使样品分散均匀,用移液枪取20µL样品均匀涂抹在氧化锡(ITO)导电玻璃上,涂覆面积为1 cm×1 cm。自然晾干后放置在烘箱中80℃下干燥2 h以上。光电流测试的电解质溶液为0.5 mol·L-1Na2SO4,工作电压为-0.2 V,使用光源为300 W氙灯(λ≥420 nm),光源开闭间隔20 s进行光电流信号采集。电化学测试包含Mott-Schottky曲线和Nyquist曲线测试。Mott-Schottky曲线:电解液为0.5 mol·L-1Na2SO4,使用频率为 1、1.5 和 2 kHz。Nyquist曲线:电解液为 5×10-3mol·L-1[K3Fe(CN)6]/[K2Fe(CN)6](物质的量之比 1∶1)和 0.1 mol·L-1KCl混合溶液,频率范围为0.1~105kHz。
1.5 光解水产氢测试
采用LabSolar-Ⅲ AG系统测定光催化剂的光解水产氢性能。在Pyrex反应器内加入50 mg催化剂、90 mL水和10 mL牺牲剂三乙醇胺,同时加入75µL质量分数2%的氯铂酸溶液(按Pt计)为助催化剂。经300 W氙灯(λ≥420 nm)光照,反应溶液温度15℃,搅拌速率400 r·min-1。产生的H2由带有热导检测器的GC-7900型气相色谱仪在线分析,每隔1 h记录一次氢气的量。光解水产氢稳定性测试:在12 h内测试CNNS-30在可见光下分解水产氢速率(HER),测试时每隔4 h抽真空以保证测量准确性。
2 结果与讨论
CNNS-x的合成路线如图1所示。首先,将尿素和TAPD混合均匀。在热聚过程中,尿素先缩聚成三聚氰酸[17],所生成的三聚氰酸易与TAPD通过氢键形成超分子[18-19];最后,进一步高温缩聚成CNNS-x。

图1 CNNS-x的合成路线示意图Fig.1 Synthetic route illustration of CNNS-x
2.1 CNNS-x的形貌和结构
首先,我们分别用FESEM、TEM和全自动独立多站比表面积和孔隙度分析仪对所得产物的形貌和比表面积进行表征。图2A、2B分别为掺入30 mg TAPD所得到的CNNS-30的FESEM和TEM图。由图可见,所得CNNS-30的形貌为二维片层结构,与以尿素作为前驱物制得的CNNS(图2C、2D)相比,CNNS-30纳米片相对较厚,径向尺寸较小。图3为CNNS-x的氮气吸附-脱附等温线,所得产物的比表面积用BET(Brunnauer-Emmett-Teller)公式计算。其中 CNNS 的比表面积(SBET)为 131 m2·g-1,而掺入 30 mg TAPD的CNNS-30的比表面积为92 m2·g-1。由此推断,TAPD的加入不利于氮化碳的径向聚合,从而使其比表面积减小。为此,我们还测试了其他样品的比表面积,如表1所示,CNNS-10、CNNS-50和CNNS-100 的比表面积分别为 96、69 和 53 m2·g-1。由此证明,TAPD的增加会影响氮化碳聚合生长的方向,使所得氮化碳纳米片比表面积减小,从而使催化剂表面的活性位点减小。

图2 CNNS-30和CNNS的FESEM图(A、C)和TEM图(B、D)Fig.2 FESEM images(A,C)and TEM images(B,D)of CNNS-30 and CNNS
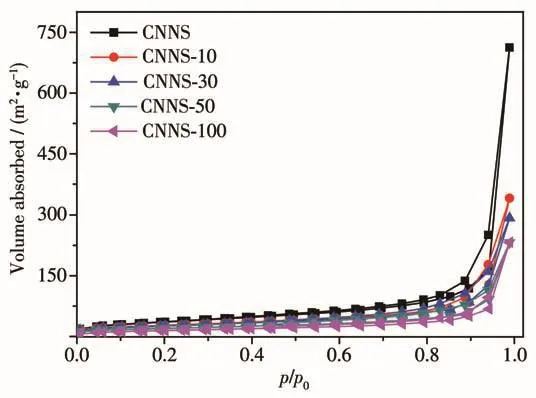
图3 CNNS-x和CNNS的氮气吸附-脱附等温线Fig.3 Nitrogen adsorption-desorption isotherms of CNNS-x and CNNS

表1 CNNS-x和CNNS的比表面积、禁带宽度及光解水产氢速率Table 1 Specific surface area,band gap and hydrogen evolution rate of CNNS-x and CNNS
随后,我们采用XRD研究CNNS-x的晶相结构,如图4所示,所有样品均出现g-C3N4的2个典型衍射峰,分别位于13.0°和27.4°处,其中,13.0°处的衍射峰来自g-C3N4的(100)晶面衍射,归因于其面内3-s-三嗪单元的有序排列;27.4°处的衍射峰是氮化碳的(002)晶面衍射,归因于氮化碳芳香族单元的层间堆积[4,20-21]。TAPD加入后,CNNS-x在13.0°的衍射峰位置基本没有变化,而在27.4°处的衍射峰向小角度方向发生偏移,并且随着TAPD加入量的增加,偏移程度逐渐增加。说明TAPD的加入对氮化碳平面的有序排列结构影响较小,但使得其层间距离增大[16]。相比CNNS,CNNS-x在13.0°和27.4°处的强度都有所减弱,这是由于嘧啶环的引入,导致氮化碳结构单元的有序性下降[22]。
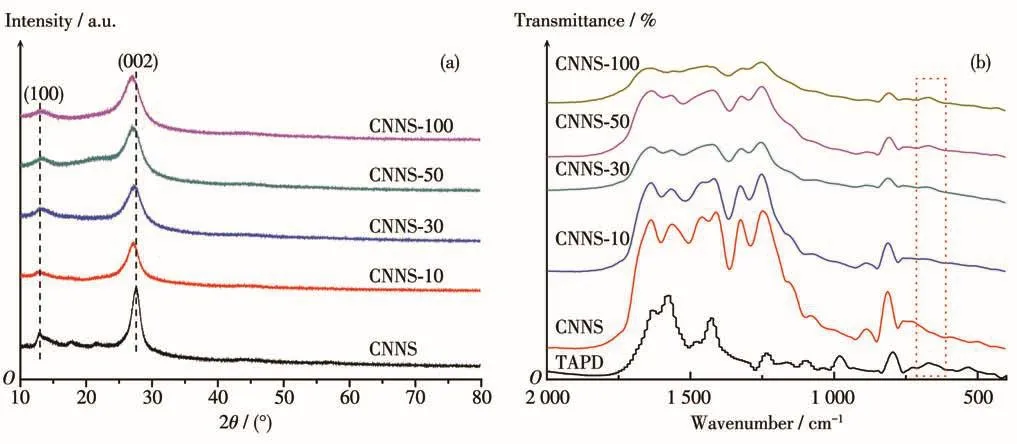
图4 (A)CNNS-x和CNNS的XRD图及(B)CNNS-x、CNNS和TAPD的FTIR图Fig.4 (A)XRD patterns of CNNS-x and CNNS,and(B)FTIR spectra of CNNS-x,CNNS and TAPD
为了进一步证明嘧啶环已成功引入氮化碳骨架结构中,我们分别采用元素分析、傅里叶红外光谱(FTIR)和X射线光电子能谱(XPS)分析CNNS-x的元素组成、官能团和价态。表2为CNNS-x和CNNS的N、C和H元素分析结果。由于TAPD与氮化碳都只含有C、N和H元素,所以无法通过元素分析来估算引入氮化碳共轭体系中TAPD的量。但由于TAPD的碳氮比(nC/nN)要高于氮化碳的碳氮比。所以可以根据所得氮化碳的碳氮比,来判断引入氮化碳共轭体系中TAPD的变化趋势。由表2可知,CNNS-x的nC/nN值都高于CNNS,并且随着TAPD添加量的增加,CNNS-x的nC/nN值逐渐增加。由此我们推测,前驱物中TAPD的添加量越多,就会有更多的TAPD分子引入到氮化碳共轭体系中。图4B为CNNS-x和CNNS的FTIR谱图。与CNNS相比,所有的CNNS-x除了在1 670~1 250和804 cm-1处出现g-C3N4典型七嗪环伸缩振动和面外弯曲振动外,在675 cm-1附近出现来自嘧啶环C—H的面外弯曲振动吸收峰,并且随着TAPD掺入量的增加,该吸收峰逐渐增强。由此说明TAPD的嘧啶环已共聚入氮化碳的π共轭体系(图1)。图5是CNNS-30和CNNS的XPS谱图。其中,图5A是CNNS-30和CNNS的全谱图,它们都有3个吸收峰(288、399和533 eV),分别对应C1s、N1s和O1s的特征峰,C和N元素来自氮化碳的骨架,少量的O元素可能是由样品吸附空气中的含氧分子如水、氧气或二氧化碳造成的。图5B是CNNS-30和CNNS的C1s高分辨谱图,二者谱峰位置基本一致,都可拟合为287.7、285.5和284.3 eV三个峰,分别归属于g-C3N4的sp2杂化碳(N—C=N,C1)、氮化碳平面边缘碳(C—NHn,C2)和石墨相碳(C—C/C=C,C3)。图5C是CNNS-30和CNNS的N1s高分辨谱图,位于398.2、399.0和400.4 eV处的峰分别对应氮化碳平面结构上的sp2杂化氮(N—C=N)、三元氮(N—(C)3)和氨基氮(C—NH)[23]。由此可见,嘧啶环的引入并未改变氮化碳的元素基本构成和价态。但值得注意的是C1s高分辨谱图(图5B)中C3与C1的峰面积比由0.28增加到0.67,进一步证明嘧啶环已被成功引入g-C3N4的共轭体系中。

表2 CNNS-x和CNNS的元素分析Table 2 Element analysis of CNNS-x and CNNS

图5 CNNS-30和CNNS的XPS谱图Fig.5 XPS spectra of CNNS-30 and CNNS
2.2 CNNS-x的吸光性能和光电行为
众所周知,通过改变氮化碳的分子结构就能实现对其能带结构的调控[11]。为此我们测试了所得产物的UV-Vis DRS谱图(图6A)和Mott-Schottky曲线(图6C)。由图6A可见,随着TAPD量的增加,CNNS-x的吸收范围逐渐向长波方向移动,同时其吸光强度也相应增强。根据对应的Tauc曲线(图6B),CNNS-x的禁带宽度(Eg)都小于CNNS的Eg;同时,当TAPD的掺入量由10 mg逐渐增加到100 mg,CNNS-x的Eg也相应由2.60 eV逐渐减小至2.38 eV。带隙的减小能促进光催化剂对可见光的利用效率。从而提高其光解水产氢效率。图6C为在2 kHz频率下所测得样品的Mott-Schottky曲线,结果表明CNNS-x的导带电位随着TAPD加入量的增加,逐渐向正向移动。因此,CNNS-x的Eg减少的部分原因是其导带变化引起的。
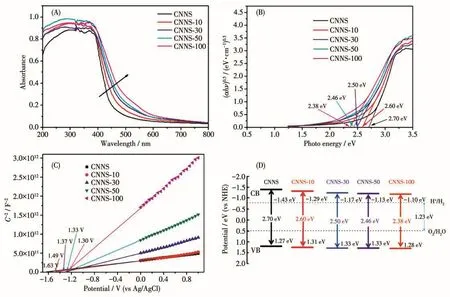
图6 CNNS-x和CNNS的UV-Vis DRS谱图(A)、对应的Tauc曲线(B)、Mott-Schottky曲线(C)和能带结构示意图(D)Fig.6 UV-Vis DRS spectra(A),corresponding Tauc plots(B),Mott-Schottky plots(C)and band structure diagram(D)of CNNS-x and CNNS
根据光解水原理,光解水产氢效率除了决定于光催化剂对光的吸收效率和催化剂表面活性位点外,还取决于光生载流子的分离效率[1,5]。为此,我们依次测定了光催化剂的Nyquist曲线、稳态荧光光谱(PL)和光电流-时间曲线(图7)。如图7A所示,在由光催化剂制成的薄膜电极的Nyquist曲线中,CNNS-30的圆弧半径电波最小,说明CNNS-30的阻抗最小,其导电性能最好[23-24]。图7B为光催化剂的稳态荧光光谱图,其中CNNS-30的荧光强度最弱,说明CNNS-30光生载流子的分离效率最高[23]。结合前面XPS的分析结果,我们有理由相信,TAPD的加入使得g-C3N4中石墨相碳(C—C/C=C)的比例增加,从而使其导电性能增加,有利于光生载流子快速到达光催化反应活性点,从而提高其光生载流子的分离效率。但过多TAPD的加入反而会使CNNS-x的阻抗增加,导电性能下降,光生载流子的分离效率下降。所制得氮化碳的光电流-时间曲线也呈现出同样的变化趋势(图7C),CNNS-30具有最强的瞬时光电流。得益于较高的吸光效率和较高的光生载流子的分离效率,CNNS-x应具有优良的可见光光催化产氢性能。

图7 CNNS-x和CNNS的Nyquist曲线(A)、稳态荧光光谱(B)和瞬时电流曲线(C)Fig.7 Nyquist plots(A),steady state PL spectra(B)and transient photocurrent(C)of CNNS-x and CNNS
2.3 CNNS-x的光解水产氢性能
图8A是样品经可见光(λ>420 nm)光照3 h后的平均产氢速率柱状图。由图可知,CNNS-x的产氢速率远大于CNNS的产氢速率。值得注意的是,TAPD加入量对CNNS-x光解水产氢速率的影响呈现先增后减的趋势。其中CNNS-30的产氢速率最高,达到了57 µmol·h-1,约为CNNS的产氢速率(14 µmol·h-1)的4倍。结合CNNS-x的能带结构(图6D)和光电行为(图7)可知,掺入TAPD所制得的CNNS-x之所以显现出优良的光解水产氢性能,一方面是由于富碳TAPD的掺入,得到了具有较窄带隙的CNNS-x,拓宽了氮化碳对可见光的吸收范围和吸收强度,提高了其对可见光的利用效率;另一方面,g-C3N4中石墨相碳(C—C/C=C)的比例增高,降低了氮化碳的阻抗,提高了光生载流子的分离效率。但TAPD的掺入会使g-C3N4的导带电位向下移动(图6D),从而使其光催化还原的效率下降。综合以上3个效率的影响,CNNS-x的光解水产氢活性随着TAPD的掺入量出现了先增后减的趋势。另外,所制得的CNNS-x具有稳定的光解水产氢活性,以CNNS-30为例,在12 h的可见光(λ>420 nm)照射下,CNNS-30的光催化活性没有出现下降的迹象(图8B)。
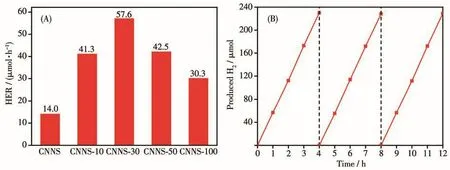
图8 (A)CNNS-x和CNNS的产氢速率及(B)CNNS-30光解水产氢稳定性测试Fig.8 (A)Hydrogen evolution rate of CNNS-x and CNNS and(B)stability test of photocatalytic H2 evolution over CNNS-30
3 结 论
以尿素为原料,掺入不同量的2,4,6-三氨基嘧啶(TAPD),通过一步热聚合反应成功得到碳自掺杂氮化碳(CNNS-x)光催化剂。形貌和结构分析显示,CNNS-x的片层结构和元素组成没有发生改变。然而,芳香族杂环的引入和芳香碳取代三嗪环中的氮原子都能有效拓展氮化的π共轭体系中的电子离域,提高光生电子的分离效率和电子的传输能力,进而提升CNNS-x的光解水产氢性能,且其具备长效性和稳定性。其中,当TAPD掺入量为30 mg时,CNNS-30的光解水产氢效率最佳,约为石墨相氮化碳纳米片的4倍。本工作为氮化碳基光催化剂的制备及光解水产氢的应用提供了思路。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
物理学报(2022年6期)2022-03-30
石油化工高等学校学报(2021年3期)2021-07-15
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
物理学报(2020年16期)2020-08-29
热处理技术与装备(2019年1期)2019-03-14
电子制作(2018年12期)2018-08-01
科技风(2018年9期)2018-05-14
