
Bartter’s syndrome:clinical findings,genetic causes and therapeutic approach
2021-04-08 05:21FlaviaCristinaCarvalhoMradlviaBouissouMoraisSoaresLuizAlbertoWanderleydeMenezesSilvaPedroVersianidosAnjosMenezesAnaCristinaSimesSilva
World Journal of Pediatrics 2021年1期
Flavia Cristina Carvalho Mrad ,2·Sílvia Bouissou Morais Soares ·Luiz Alberto Wanderley de Menezes Silva ·Pedro Versiani dos Anjos Menezes·Ana Cristina Simões-e-Silva
Abstract Backgound Bartter’s syndrome (BS) is a rare group of salt losing tubulopathies due to the impairment of transport mechanisms at the thick ascending limb of the Henle’s loop.Data sources Literature reviews and original research articles were collected from database,including PubMed and Scopus.Results According to the time of onset and symptoms,BS can be classified into antenatal and classic BS.Molecular studies have identified different subtypes of BS.BS types I,II and III are caused by mutations on genes encoding the luminal Na + —K + —2Cl − co-transporter,the luminal K+channel ROMK,and the basolateral chloride channel ClC-Kb(CLCNKB),respectively.Loss-of-function mutations of Barttin CLCNK type accessory beta subunit cause BS type IVa.Simultaneous mutations of CLCNKB and CLCNKA cause BS type IVb.BS type V consists in a novel transient form characterized by antenatal presentation due to mutations in the MAGE family member D2.Severe gain-of-function mutations of the extracellular calcium sensing receptor gene can result in an autosomal dominant condition of BS.Main clinical and biochemical alterations in BS include polyuria,dehydration,hypokalemia,hypochloremic metabolic alkalosis,hyperreninemia,high levels of prostaglandins,normal or low blood pressure,hypercalciuria and failure to thrive.Treatment focuses mainly at correcting dehydration and electrolyte disturbances and in measures to reduce polyuria,including the use of nonsteroidal anti-inflammatory medications to control excessive renal prostaglandin E2 production.Conclusions Early diagnosis and treatment of BS may prevent long-term consequences such as growth failure,nephrocalcinosis and end-stage renal disease.
Keywords Bartter’s syndrome·Hypercalciuria·Hypokalemia·Metabolic alkalosis·Nephrocalcinosis
Introduction
Bartter’s syndrome (BS),firstly described in 1962 by Frederic Bartter and collaborators is a heterogeneous group of tubulopathies with recessive and dominant autosomal inheritance due to impairment of the salt reabsorption at the thick ascending limb of Henle’s loop [1,2].It is characterized by hypokalemia,metabolic alkalosis,hyperreninemia,and secondary hyperaldosteronism,high levels of prostaglandin E2 (PGE2) with normal or low blood pressure,renal salt wasting and hyperplasia of juxtaglomerular apparatus [1,3].Nephrocalcinosis can be commonly found in patients with BS [3].Phenotypically,BS can be classified as antenatal (aBS) and classic form of BS (cBS) based exclusively on the time of onset and characteristic symptoms [4— 8].
Very few epidemiological data are available regarding the frequency of BS,but it is generally considered a rare disease.In a report from the Framingham Heart Study,the prevalence of BS was 1 per 1,000,000 live births [9].
The aim of this review was to summarize pathophysiology and clinical findings and to describe recent data on the genes-related to BS and the therapeutic approach.
Pathophysiology
The thick ascending limb (TAL) of the loop of Henle is responsible for the reabsorption of nearly 20% of filtered sodium [4].Sodium (Na+),potassium (K+) and chloride(Cl−) are co-transported through the apical membrane of the TAL via the transporter named NKCC2.NKCC2 is a furosemide-sensitive Na+—K+—2Cl−cotransporter encoded by the solute carrier family 12 member 1 (SLC12A1) gene[7,9].Na+is actively pumped out of the TAL cell by basolateral Na+—K+—adenosine triphosphate (Na+/K+-ATPase),whereas Cl−leaves the cell through specific Cl−channels,which are also located at basolateral membrane,and termed Cl−channel Ka (CLC-Ka) and Cl−channel Kb(CLC-Kb) [9].Both of these chloride channels require the protein Barttin as a subunit to exert proper function [10](Fig.1).
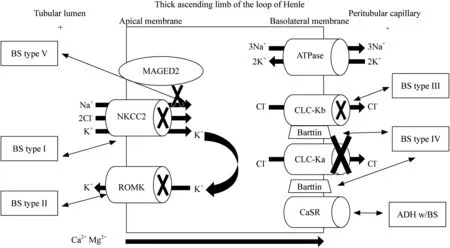
Fig.1 Mechanisms of transport at thick ascending limb of the Henle’s loop.Transport of NaCl at the thick ascending limb of the loop of Henle occurs via the apical membrane by NKCC2,a furosemidesensitive Na + —K + —2Cl − co-transporter.NKCC2 activity is maintained by the electrochemical gradient created by the basolateral Na +/K + -ATPase,which is also regulated by MAGED2 and supported by the recycling of K + via ROMK.The lumen transepithelial voltage is positive due to the passive transport of Cl − by means of ClC-Ka and ClC-Kb basolateral channels and also because of the K + recycling.The lumen positive voltage stimulates passive paracellular transport of Mg 2+ and Ca 2+ .CASR promotes calcium reabsorption in response to increases of its plasma concentration.NaCl sodium chloride,K +potassium,Na + sodium,Cl − chloride,Ca 2+ calcium,Mg 2+ magnesium,NKCC2 sodium-chloride cotransporter,Na + /K + -ATPase sodium—potassium-adenosine triphosphate,ROMK medullary potassium channel,ClC-Ka type a chloride channel,ClC-Kb type b chloride channel,CASR calcium-sensing receptor,MAGED2 mage family member D2
The K+is recycled into the lumen via ROMK (renal outer medullary K+channel) [11],encoded by the potassium voltage-gated channel subfamily J member (KCNJ1)gene;otherwise,it would become the rate-limiting step for reabsorption of NaCl.K+recycling determines a lumenpositive transepithelial voltage,which promotes passive paracellular transport of Na+,K+,magnesium (Mg+2) and calcium (Ca+2) in this tubular segment [7,8](Fig.1).
Chronic Na+depletion leads to a contraction of the extracellular volume.Hypovolemia results in the activation of the renin—angiotensin—aldosterone system.Secondary hyperaldosteronism determines increasing of the Na+reabsorption,K+secretion and hydrogen (H+) secretion by activating of the Na+/K+-ATPase pump [4,7,10,11].Salt depletion and local release of angiotensin II (Ang II) increase cyclooxygenase-2 (COX-2) local activity,which,in turn,induces excessive production of PGE2 [4,6].Whereas Ang II produces vasoconstriction of efferent arteriole,PGE2 determines vasodilation of afferent arteriole that worsens urinary loss of Na+by increasing glomerular filtration pressure,leading to a glomerular hyperfiltration [4,7,8].Hyperreninism may not always be caused by hypovolemia.Because of the irregular tubuloglomerular feedback due to macula densa cells mutations,renin secretion happens irrespective of volume status[12].Reduced NaCl reabsorption combined with glomerular hyperfiltration results in the main clinical alterations of BS[2,4,10].Polyuria is usually present due to the inability to generate an appropriate medullary interstitial tonicity [12].
A complete diagram showing the pathophysiology of BS is presented in Fig.2.
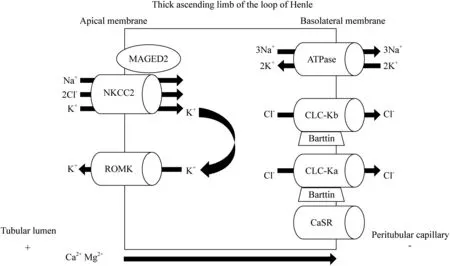
Fig.2 Defects of renal channels and/or of transport mechanisms in the subtypes of Bartter’s syndrome.Na + is electrically reabsorbed along with K + and 2 Cl − through NKCC2 (BS type I defect).K + is recycled through ROMK (BS type II defect) to provide regular supply of K + to the lumen otherwise the NaCl reabsorption would be limited.Cl − exits the tubular cell through ClC-Kb (BS type III defect),including barttin subunit (BS type IV defect).The upper-regulated function of CaSR inhibits NKCC2,resulting in autosomal dominant hypocal-cemia with BS.MAGED2 mutations affect NKCC2 regular expression and function (BS type V defect).BS Bartter’s syndrome,K + potassium,Na + sodium,Cl − chloride,Ca 2+ calcium,Mg 2+ magnesium,NKCC2 sodium-chloride cotransporter,Na + /K + -ATPase sodium—potassiumadenosine triphosphate,ROMK medullary potassium channel,ClC-Ka type a chloride channel,ClC-Kb type b chloride channel,CaSR calcium-sensing receptor,ADH w/BS autossomal dominant hypocalcemia with BS,MAGED2 mage family member D2
Genetic causes
Loss-of-function mutations of several genes encoding transporters related to NaCl reabsorption at the TAL cause at least six different subtypes of BS [4,7].
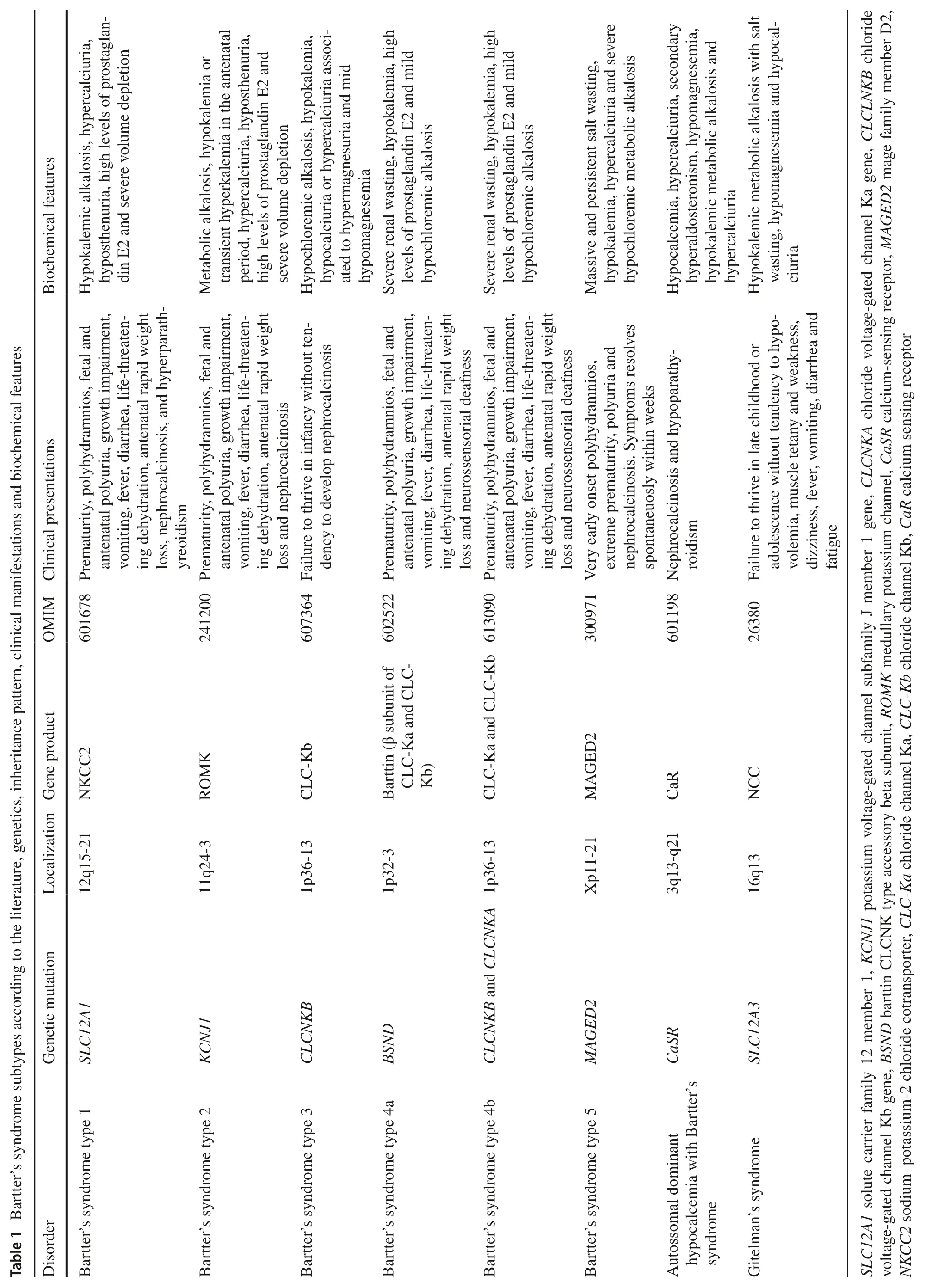
BS type I (OMIM 601678) is caused by mutations in theSLC12A1gene that encodesNKCC2[4,13],being responsible for the majority of NaCl reabsorption in the apical membrane of epithelial cells at the TAL.SLC12A1couples with its memberSLC12A2to mediate actively the electroneutral transportation of 1 Na+,1 K+and 2 Cl−through the cell membrane.The result ofSLC12A1disruption is hypercalciuria and severe volume depletion with early presentation in BS patients [13].AlthoughNKCC2is primarily regulated by cyclic adenosine monophosphate,the regulation of this cotransporter is complex.Some recently in vitro studies have shown that this cotransporter receives some limited,but positive,regulation from with-no-lisine kinase 3,which hypothetically works as a Cl−sensor [13— 17].Furthermore,Ang II has a main role in the regulation ofNKCC2by triggering its activation and consequently impacting on Na+and fluid homeostasis at the distal nephron.NKCC2is expressed on the apical membrane of TAL epithelial cells and blocked by loop diuretics [15— 20].Hypokalemia is result of a increased aldosterone level,which increases the activity of epithelial Na+channel (ENaC) in the distal convoluted tubule,resulting in a increased Na+reabsorption and K+excretion by ROMK channel [7,8,20].Hypercalciuria is the result of NKCC2 defective function,which increases intraluminal concentration of Cl−and produces electronegative gradient that prevents the paracellular reabsorption of positive ions such as Ca+2[20].Recently,whole exome sequencing revealed novel pathogenic mutations in theSLC12A1gene in patients with BS type I with varying degrees of hypercalcemia and primary hyperparathyroidism,constituting atypical presentations [14,21,22].Han et al.[23]identified 11 novelSLC12A1variants and suggested that the aberrant exon skipping is a previously unrecognized mechanism by which an exonic variant inSLC12A1can lead to BS type I.
BS type II (OMIM 241200) results from mutations in theKCNJ1gene that encodes ROMK [2,3].KCNJ1interacts with WNK kinases,Src family protein tyrosine kinase,plenty of SH3,and protein kinase C to regulate renal K+handling.Mutations inKCNJ1affecting exon 2 of the gene,that are usually missense/nonsense,results in loss of K+channel activity through the phosphorylation sites alteration,or through the frameshift the gene open reading frame [13,24].ROMK activity is required for proper NKCC2 function,thus explaining the fact that BS type I and type II have similar prenatal presentation,which is characterized by hypercalciuria [4].Khandelwal et al.[25]described a 14-year-old patient with late-onset BS type II,who was diagnosed after assessment for isolated nephrocalcinosis due to hypercalciuria.This case emphasizes the importance in screening theKNJ1gene in patients with hypercalciuria and nephrocalcinosis,regardless of age.
BS type III (OMIM 607364) or cBS is caused by mutations Cl−voltage-gated channel Kb (CLCNKB)gene,which encodes CLC-Kb [14]and cause loss of function and consequently impairment in the reabsorption of Cl−at the TAL[12].The functional severity ofCLCNKBgene mutations is an important determinant of the broad cBS phenotype,since the whole gene deletion variants may lead to a more severe phenotype of patients [16,17,26].An investigation regarding the consequences of 9 mutations in theCLCNKBrevealed that at least three different classes of mutations can interfere partly or totally with the intracellular compartment of the channel or even directly affects the membrane surface,resulting in a large range of alterations in channel gating properties [27].Heterogeneous cohorts showed that the homozygous deletion encompassing the entireCLCNKBgene represented the most common molecular finding in BS type III with classic presentation,which is frequently associated with most severe phenotype and hypercalciuria [15,16].
BS type IVa (OMIM 602522) is caused by mutations in the Barttin CLCNK type accessory beta subunit gene,which encodes the protein Barttin,which is present in CLC-Ka and CLC-Kb channels,both located at the TAL,and in the stria vacularis at the inner ear,characterized by severe renal salt wasting and sensorineural deafness [4,13,28— 30].
BS type IVb (OMIM 613090) is caused by dysgenic mutations inCLCNKBandCLCNKAgenes,which encodes CLC-Kb and CLC-Ka channels,respectively [4,13].A study investigating the depletion of a Barttin enzyme,DHHC-type containing 7 (DHHC7),in mices suggested that this protein might play an important role in chloride channel dysfunctions [31].The affected mices,when fed with a low salt diet,developed hyponatremia and mild metabolic alkalosis,which are very common metabolic alterations of human BS type IVb [31].
BS type V (OMIM 300971),previously described as a transient form of BS (tBS),was firstly reported in 2016 and it is caused by mutations ofMAGED2X-linked gene [7].In developing and adult kidneys,MAGED2is expressed in the TAL and the distal renal tubules,being essential for fetal renal salt reabsorption and amniotic fluid homeostasis.MAGED2is also critical for the maintenance of a normal pregnancy regarding the time of gestation,since the fetal genotype is both necessary and sufficient for the full obstetrical and perinatal phenotype,as indicated by Laghmani et al.[6,7]results.NKCC2andNCC,sodium chloride cotransporters of fetal tubular cells,are both affected by mutations in this X-linked gene,since current data indicates thatMAGED2promotes the biogenesis of these cotransporters.[6,7,32].The simultaneous reduction in the expression ofNKCC2andNCCmay explain,therefore,at least in part,the severity of BS type V features in patients withMAGED2mutations [7,32].
Autosomal dominant hypocalcemia with BS (OMIM 601198),caused by heterozygous mutation in theCaSRgene,is not associated with antenatal BS.CaSRgene plays a key role in the regulation of calcium-phosphate metabolism by controlling parathyroid hormone secretion and the rate of calcium reabsorption by the kidney in response to variations in serum calcium levels.The autosomal dominant hypocalcemia with BS (OMIM 601198) is due to a gainfunction mutation of theCaSRgene that encodes the Ca2+sensing receptor at the basolateral membrane of TAL cells.The increased activityCaSRgene inhibits NKCC2 function by directly targeting its phosphorylation and activation pathway [2,7,13,24,33].
Clinical and biochemical findings
In the aBS symptoms usually starts before birth with the development of polyhydramnios between 24 and 30 weeks of gestation,intrauterine growth restriction,fetal polyuria [10,34— 36].After birth,usually premature,clinical and laboratory findings of aBS include hyposthenuria,intense salt wasting,polyuria,high plasma level of renin with normal to low blood pressure,hypokalemia,moderate hypochloremic metabolic alkalosis and rapid weight loss [13,35,36].Disruption of ROMK channel may also lead to transient hyperkalemia in the neonatal period [35].PGE2 levels are high in blood and urine as a secondary phenomenon,leading to vomiting,fever,diarrhea and also glomerular hyperfiltration [4,8,13,36,37].Urinary common findings in BS patients include persistently elevated urinary chloride concentrations and increased urinary loss of Na+,K+and Ca2+,which usually help clinical differentiation of BS from other causes of metabolic alkalosis[3,13,35,36].
BS can be prenatally suspected by amniotic fluid biochemistry analysis,allowing appropriate management before and after birth [38,39].Garnier et al.[38]proposed a method known as Bartter’s index (total protein and alphafetoprotein multiplication),which is usually significantly lower in amniotic fluid in patients with aBS (94.3% sensitivity and 100% specificity).
Infants may exhibit difficulty feeding,growth impairment,polydipsia,polyuria,life-threatening dehydration and severe electrolyte imbalance associated with hypochloremic metabolic alkalosis [2,35,36].Fatigue,dizziness,weakness,muscle cramps due to chronic hypokalemia may occur in older children [35,36].Ultrasonography of the kidneys detects bilateral medullary nephrocalcinosis,which can be observed after some weeks of severe hypercalciuria [38].In some cases,characteristic facial aspect including triangularshaped face with a prominent forehead,large eyes,protruding ears and drooping mouth may be observed [35,36,40].
cBS has a significant phenotype variability,ranging from hypocalciuria to hypercalciuria associated to hypermagnesuria,without the tendency to develop nephrocalcinosis [16,17,36].Clinical symptoms and biochemical markers of Gitelman’s syndrome and cBS may overlap and thus genetic analysis is required to make an accurate diagnosis.Gitelman’s syndrome is a rare salt-losing tubulopathy characterized by hypokalemic metabolic alkalosis with hypomagnesemia and hypocalciuria.The disease is recessive inherited and caused by inactivating mutations in theSLC12A3gene that encodes the NCC [41].
BS type V,caused byMAGED2mutations,is clinically characterized by a very early onset of severe polyhydramnios,extreme prematurity,massive and persistent salt wasting and polyuria,leading to high mortality [5,6,32].After birth,which is usually premature,the surviving patients tend to present hypokalemia,a more severe hypochloremic metabolic alkalosis and hypercalciuria that can lead to nephrocalcinosis.The symptoms in patients withMAGED2mutations resolves spontaneously,within weeks,with no further explanation for this transient nature of the phenotype [6,32].
Patients with autosomal dominant hypocalcemia with BS also present decreased chloride reabsorption,negative balance of sodium chloride,secondary hyperaldosteronism,hypomagnesemia,and hypokalemic metabolic alkalosis.Patients withCaSRgene mutation have a tendency to develop abnormal calcium metabolism,which may lead to hypercalciuria,nephrocalcinosis,and hypoparathyroidism.Some patients may develop asymptomatic hypocalcemia.[7,13,33,42].
Renal biopsy is usually not required in BS juxtaglomerular hyperplasia represents a characteristic finding,but not necessary for diagnosis [43].Focal segmental glomerulosclerosis may be secondary to the adaptive response to chronic hyperfiltration,due to continuous salt loss and stimulation by the renin-angiotensin system,resulting in a risk of progression to end-stage renal disease [44]
Although rare,BS can occur in adults and during pregnancy due to phenotypic variations.These patients may present hypokalemia,metabolic alkalosis,high excretion of K+and Cl−in the urine,high serum levels of renin and aldosterone,hypercalciuria,nephrocalcinosis and normal or reduced glomerular filtration rate [45— 47]
Table 1 shows the main genetic defects responsible for different subtypes of BS and their associated clinical manifestations and biochemical features.
Genetic investigation
Analysis of the genomic DNA mutations will identify the fundamental defect in BS and is considered very important due to clinical and genetic heterogeneity characteristics of this syndrome [2,9,36].Genetic analysis can also use DNA extracted from cultured amniocytes obtained by amniocentesis [35].
Genetic testing can specify or even change the clinical diagnosis,leading to more tailored treatment.Besides,specific genetic counseling and screening for at-risk relatives can be performed [2,48— 50].The whole-exome/genome sequencing is mostly used as a first-line approach.This is explained by the fact that targeted panel relies on the accuracy of the clinical diagnosis if genetic testing is performed by means of the sequencing of single genes.This would lead to a tailored approach for achieving the genetic diagnosis [49].The most frequent mutations among BS phenotypes are those affecting theKCNJ1gene(ROMK channel),followed by theSLC12A1gene (NKCC2 channel),CLCNKBgene (ClC-Kb channel),BSNDgene(barttin protein),MAGED2gene (MAGED2 protein) andCaSRgene (calcium sensing receptor protein) [13,20,48].
Genetic investigation is fundamental for differential diagnosis between cBS and Gitelman’s syndrome [4,44].Detection of biallelic inactivating mutations inSLC12A3confirms Gitelman’s syndrome diagnosis [41,51].
However,it is worth mentioning that in about one-third of patients with pediatric-onset tubulopathy,a genetic diagnosis cannot be established.The same is true for twothirds of patients with adult onset BS [48— 50].
Treatment
Treatment includes lifelong fluid and electrolyte supplementation,and can include aldosterone antagonist or even the use of nonsteroidal anti-inflammatory drugs(NSAIDs) to inhibit excessive PGE2.However,the safety of long-term treatment with NSAIDs,especially in preterm infants,is a subject of controversy [5,35,52].
High levels of PGE2 are due to the activation of COX-2 in macula densa.Therefore,COX-2 inhibition by indomethacin is widely used for the treatment of BS in pediatric patients [35,53].The reduction of glomerular hyperfiltration is necessary,since this condition is associated to focal glomeruloscleroris occurrence and,consequently,interstitial fibrosis [54].The recommended dose of indomethacin is 1—5 mg/kg/day in two or three divided doses[35].Indomethacin has side effects on gastrointestinal tract,emphasizing the importance of careful monitoring[35,52].The use of COX-2 selective inhibitor as rofecoxib has suppressed hyperreninemia to an equal level as indomethacin and probably with less gastric side effects [55].However,a high risk of cardiovascular events in patients receiving COX-2 selective inhibitors has limited its use[56].It is worth mentioning that the antenatal administration of indomethacin to prevent prematurity and to reduce polyhydramnios is controversial,since it may increase the risk of necrotizing enterocolitis,affect the maturation of the kidneys,the total number of nephrons in the fetus and may also produce premature closure of the ductus arteriosus [35,39,45,52].
Besides the use of potassium supplements,potassiumsparing diuretics,such as spironolactone (aldosteroneblocking receptor) or amiloride (a direct inhibitor of the ENaC channel),may help elevating serum potassium and reverse metabolic alkalosis.However,thid treatment requires caution,since treatment with potassium-sparing diuretics may be dangerous in situations of gross salt and water loss and contraction of the circulatory volume [2,35,52].
Angiotensin-converting-enzyme inhibitors and angiotensin receptor blockers can decrease the high levels and/or effects of Ang II and aldosterone,thus controlling proteinuria,and increasing serum potassium concentration in some cases [57].Because polyuria and dehydration are common symptoms,caution is necessary due to the potential risk of acute kidney injury in this high-risk group of patients [52,58].
A recent randomized clinical trial showed a beneficial effect by adding acetazolamide (AZM) to the standard therapy of BS.In this trial,the use of AZM reduced serum bicarbonate and increased serum potassium levels.In addition,AZM also decreased serum levels of aldosterone and plasma renin activity.Although no specific AZM related effects were identified during the short-period trial,long-term efficacy and safety require further investigation,since this medication is not recommended for patients with impaired renal function [59].
Osteopenia is an established finding in BS,probably due to the hypercalciuric phenotype.Therefore,bone mineral density must be performed and monitored in such patients[53].The stature reached by children with BS,even when under adequate treatment,is usually considered short for the age.In those cases,growth hormone therapy has been successful for the treatment of growth retardation and short stature potentially associated with BS.However,previous studies showed that growth hormone (GH) does not stimulate growth when severe hypokalemia is present.Indeed,GH should only be used if growth retardation still occurs in a spite of appropriate administration of fluid and electrolytes[52,60].
It must also be mentioned that,under stressful situations,including acute illness,surgical procedures and trauma,the levels of blood electrolytes may rapidly change,requiring immediate and vigorous intravenous treatment .[39].
Conclusions
BS is a heterogeneous group of tubulopathies basically due to salt reabsorption impairment in the TAL.Any premature neonate that had unexplained polyhydramnios and any child with growth failure,hypokalemia,and hypochloremic metabolic alkalosis should be investigated for BS.Correct diagnosis and early treatment improve the quality of life of these children,allowing them to reach their full growth potential.In addition,treatment may also prevent the development of complications and the impairment of renal function.It is important to emphasize that genetic investigation in children with symptoms suggestive of BS helps to establish a specific diagnosis and genetic counseling for family members.
Author contributationsAll authors have participated in the concept and design,analysis and interpretation of data;drafting or revising of the manuscript;and have approved the manuscript as submitted.
FundingNone.
Compliance with ethical standards
Ethical approvalNot needed.
Conflict of interestNo financial or nonfinancial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
 World Journal of Pediatrics2021年1期
World Journal of Pediatrics2021年1期
- World Journal of Pediatrics的其它文章
- Assessment of left-ventricular diastolic function in pediatric intensive-care patients:a review of parameters and indications compared with those for adults
- Dent disease:classification,heterogeneity and diagnosis
- Advance in the understanding of vasovagal syncope in children and adolescents
- Tribute to reviewers (January 1,2020 to December 31,2020)
- Instructions for Authors
- Intestinal microbiota and juvenile idiopathic arthritis:current understanding and future prospective