
Expert consensus of the Chinese Association for the Study of Pain on ion channel drugs for neuropathic pain
2021-04-08 08:49HongXiaoKeMaDongHuangXianGuoLiuTangHuaLiuQingLiuGuangZhaoLiuTaoSongWeiTaoDaShengWuYunXiaWangXiaoQiuYangXiaoMeiZhangHuiLiuYanQingLiu
World Journal of Clinical Cases 2021年9期
Hong Xiao, Ke Ma, Dong Huang, Xian-Guo Liu, Tang-Hua Liu, Qing Liu, Guang-Zhao Liu, Tao Song, Wei Tao,Da-Sheng Wu, Yun-Xia Wang, Xiao-Qiu Yang, Xiao-Mei Zhang, Hui Liu, Yan-Qing Liu
Hong Xiao, Hui Liu, Department of Algology, West China Hospital, Sichuan University,Chengdu 610041, Sichuan Province, China
Ke Ma, Department of Algology, Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200092, China
Dong Huang, Department of Algology, The Third Xiangya Hospital of Central South University, Changsha 410013, Hunan Province, China
Xian-Guo Liu, Department of Physiology and Pain Research Center, Sun Yat-Sen University,Guangzhou 510080, Guangdong Province, China
Tang-Hua Liu, Department of Algology, The Third People's Hospital of Yibin, Yibin 644000,Sichuan Province, China
Qing Liu, Department of Algology, The Affiliated T.C.M Hospital of Southwest Medical University, Luzhou 646000, Sichuan Province, China
Guang-Zhao Liu, Department of Algology, The Second Hospital of Hebei Medical University,Shijiazhuang 050000, Hebei Province, China
Tao Song, Department of Algology, The First Affiliated Hospital of China Medical University,Shenyang 110001, Liaoning Province, China
Wei Tao, Department of Neurosurgery, Shenzhen University General Hospital, Shenzhen 518055, Guangdong Province, China
Da-Sheng Wu, Department of Algology, The People's Hospital of Jilin Province, Changchun 130021, Jilin Province, China
Yun-Xia Wang, Department of Algology, Hubei Third People’s Hospital of Jianghan University,Wuhan 430033, Hubei Province, China
Xiao-Qiu Yang, Department of Algology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China
Xiao-Mei Zhang, Department of Algology, The First Affiliated Hospital of Kunming Medical University, Kunming 650032, Yunnan Province, China
Yan-Qing Liu, Department of Algology, Beijing Tiantan Hospital, Capital Medical University,Beijing 100070, China
Abstract Neuropathic pain (NPP) is a kind of pain caused by disease or damage impacting the somatosensory system. Ion channel drugs are the main treatment for NPP;however, their irregular usage leads to unsatisfactory pain relief. To regulate the treatment of NPP with ion channel drugs in clinical practice, the Chinese Association for the Study of Pain organized first-line pain management experts from China to write an expert consensus as the reference for the use of ion channels drugs . Here, we reviewed the mechanism and characteristics of sodium and calcium channel drugs, and developed recommendations for the therapeutic principles and clinical practice for carbamazepine, oxcarbazepine, lidocaine,bulleyaconitine A, pregabalin, and gabapentin. We hope this guideline provides guidance to clinicians and patients on the use of ion channel drugs for the management of NPP.
Key Words: Ion channel drug; Neuropathic pain; Expert consensus; Guideline; Gabapentin;Carbamazepine; Oxcarbazepine; Lidocaine; Bulleyaconitine A; Pregabalin
INTRODUCTION
Objective and significance
Neuropathic pain (NPP) is a kind of pain caused by disease or damage impacting the somatosensory system[1]; it has a complex pathogenesis. The prevalence of NPP in the general population is as high as 8.0%[2]. Based on these data, there are about 90 million NPP patients in China, and the incidence of NPP is increasing gradually.
Currently, the main treatment for NPP is clinical drug therapy, of which the most common drugs include anticonvulsants, antidepressants, opioid analgesics, and Nmethyl-D-aspartate (NMDA) antagonists. Most are ion channel drugs, but the response rate is only about 46.3%[3]. Due to the complexity of NPP etiology/pathogenesis, diverse nature of pain, duration of disease and pain ranging, a reasonable combination therapy of different ion channel drugs is an effective way to improve the clinical efficacy currently. To regulate treatment of NPP with ion channel drugs in clinical practice, the Chinese Association for the Study of Pain (CASP)organized first-line pain management experts from China to write an expert consensus as a reference for the use of ion channel drugs.
Abnormal expression of ion channels and NPP
The clinical manifestation of NPP includes decreased pain threshold, increased pain response and spontaneous pain, which are mainly related to peripheral and central sensitization. Peripheral sensitization refers to the abnormal expression of voltagedependent ion channels in primary afferent neurons, leading to increased excitability and increased pain signals. The central one refers to the continuous enhancement of synaptic transmission efficiency on the pain pathway, then amplify the pain signals.The abnormal expression of ion channels induces NPP. There are currently two types of ion channel drugs in clinical use: sodium channel blockers and calcium channel modulators. Sodium channel blockers are represented by carbamazepine, lidocaine,and BLA. They block different types of voltage-dependent sodium channels and inhibit overexcited sensory neurons. Calcium channel adjusting section comprises gabapentin and pregabalin, which selectively inhibit synaptic transmission of pain, to alleviate NPP without directly blocking effects on the calcium channel. The α2δ1 subunit located in the presynaptic calcium channel of the spinal dorsal horn is upregulated during NPP, resulting in increased release of neurotransmitters and enhanced synaptic transmission of pain. Gabapentin and pregabalin bind to this subunit, downregulating it and exerting analgesic effects. In short, by inhibiting the peripheral sensitization and central sensitization, ion channel drugs affect NPP.Sodium channel blockers and calcium channel modulators have anticonvulsant effects as well.
Treatment principle of ion channel drugs
(1) Individualized therapy: It is necessary to pay attention to the wide dose difference to achieve the effective analgesia of the patient. When a drug is not effective, the resistance occurs or effective duration shortened after long-term treatment, it is not advisable to change the drug easily. It is advised to increase the dose to obtain satisfactory results without serious side effects, but care should be taken to avoid exceeding the toxic threshold; (2) Timely administration: Take drugs with proper interval based on their onset time, to maximize analgesia effect; (3) Oral administration: Choose oral, non-invasive approach medicine as possible; (4) Closely observe the medication’s onset time, effective duration, degree of analgesic effect, and side effects to ensure an adequate course of treatment; and (5) Adjust the dosage of drugs according to the state of illness: Combination therapy should be considered when the effect of the single drug is not effective.
CLASSIFICATION AND INTRODUCTION OF ION CHANNEL DRUGS
Sodium channel antagonist agent
Mechanism and characteristics of sodium channel drugs:The sodium ion channel is a transmembrane glycoprotein on the cell membrane, which selectively allows sodium to pass through the membrane. Sodium channel antagonist agents can eliminate or alleviate acute pain, inflammatory pain, and NPP and effectively improve the symptoms of hyperalgesia of NPP[4].
Typical drugs of sodium channel antagonists:Representative drugs for sodium ion channel antagonists include carbamazepine, oxcarbazepine, lidocaine, and BLA.Carbamazepine works by inhibiting cell membrane sodium ion channels, reducing neurotransmitter release, and reducing nerve cell excitability. In addition, it also acts on gamma-aminobutyric acid (GABA) receptors, interferes with glutamate functions through NMDA receptors, and regulates central sensitization. The mechanism of oxcarbazepine is by blocking voltage-dependent sodium channels in the brain,stabilizing the neuronal cell membrane, inhibiting repetitive firing of neurons,reducing the release of synaptic impulses, and reducing high voltage-activated calcium currents in the striatum and cortical neurons, thereby reducing the glutamateinduced transmission of cortical striatum synapses[5]. Both mechanisms may be involved in the treatment of neuralgia by oxcarbazepine. Some studies have also suggested that oxcarbazepine may play a role by inhibiting substance-P mediated pain transmission[6].
Lidocaine is a typical sodium channel antagonist; it inhibits sodium ion channels and blocks the action of increased excitability of central neuron, thereby impacting peripheral and central endings and having analgesic effects[7].
BLA can state dependently inhibit voltage-gated sodium channels. Second, BLA may have anti-inflammatory and analgesic effects through many methods including lowering serum prostaglandin E2 levels, regulating serotonin (5-hydroxytryptamine)levels in the brain and stimulating the expression of dynorphin A in spinal microglia,relieving beta- endorphin inhibition which raises pain thresholds.
Pharmacokinetics, pharmacodynamics, and physicochemical properties of sodium ion channel antagonists:Carbamazepine is commonly used as anticonvulsant drugs.Its gastrointestinal absorption is slow and irregular. Due to dose-dependent induction by itself or other enzymes as well as age, comorbidities, combination therapy,etc., it appears to have high variance of individual pharmacokinetics (PKs)[8]. The formulation and food intake do not affect the rate and speed of absorption. After taking a single dose of carbamazepine, peak plasma concentration reached within 12 h, peak concentration reached 4-5 h (0.5-25 μg/mL) after oral administration of 400 mg, and steady-state plasma concentration was reached within 1-2 wk. It has been recommended that the carbamazepine plasma concentration of 4-12 μg/mL is“effective” in adults[9]. Carbamazepine is metabolized by the liver. Epoxidization is its major metabolic pathways, and it is metabolized by cytochrome P450 3A4 to pharmacologically active 10, 11- epoxy carbamazepine. Protein binding rates of these two substances are 76% and 48%-53%. They can pass through the placental barrier and be secreted with lactating. Carbamazepine’st1/2is related to the duration of treatment,25-65 h for a single administration, and 12-17 h for long-term administration[10]. Its half-life of children is significantly shortened, and there is no change in the PKs in elderly patients. PK data in patients with liver or renal disease are still insufficient(Table 1).
Oxcarbazepine is a new type of anticonvulsant drug. It is a 10-keto derivative of carbamazepine. It mainly works through its active metabolite, 10- hydroxycarbamazepine (MHD). After taking a single dose of oxcarbazepine, the absorption time is 1 to 3 h, the peak serum concentration of its metabolite MHD is 4 h to 12 h, the protein binding rate of oxcarbazepine and MHD is 60% and 40%, and thet1/2respectively is 1 to 5 h and 9 to 11 h[11]. Oxcarbazepine can be directly converted into the active metabolite MHD without P450 enzyme; therefore, the adverse effects of drugs and the drug interactions when combined are less. Oxcarbazepine and carbamazepine have different metabolic pathways in the liverviacytochrome P450 oxidase. Oxcarbazepine is reduced to MHD in the cytoplasm by reductase. MHD is not metabolized by the liver but excreted by the kidney. MHD’s steady-state levels can be reached within 2-3 d. Without liver drug enzyme induction and self-induction phenomenon and the blood drug concentration is nearly linear, making it easier to control the dose (Table 1).
The PKs of intravenous administration and transdermal lidocaine patch are very similar. About 70% of lidocaine binds to plasma proteins, mainly to α-1-acid glycoprotein. Lidocaine binds to plasma proteins in a concentration-dependent manner. It may pass through the placenta and the blood-brain barrier through passive diffusion. Lidocaine can be rapidly metabolized into a variety of metabolites in the liver with a bioavailability of about 35%. Metabolites include monomethyl glycerol dimethylaniline and glycine dimethylaniline. Their pharmacological effects are similar with lidocaine, but their activity is lower than lidocaine. Lidocaine and its metabolites are excreted by the kidneys. Less than 10% of lidocaine is excreted as its original form.When administered intravenously, lidocaine plasma clearance half-life is 81-149 min.Systemic clearance rate is 0.33-0.90 L/min (Table 1).
BLA classification is a diterpene diester, is a state-dependent sodium channel antagonist, no resistance and addiction, less adverse effects, is a potent analgesic and anti-inflammation agent. There are no studies of human PKs. After intravenous injection in rats and the blood drug-time curve appears to be an open threecompartment model. Its three-phase half-lives are: the fast-distribution phase half-life (t1/2fast-distribution) = 2.87 min, distribution phase half-life (t1/2distribution) = 11.6 min, elimination phase half-life (t1/2elimination) = 5 h. In tissues, the concentrations in the liver and adrenal glands are highest, followed by the kidney, lung, spleen, and heart. The concentration in brain is lower, while in the brainstem is higher than in the cortex. After 4 h later of administration, the concentration in each organ was reduced by 50%. Within 6 d after administration, the amount of urine excretion accounted for 46% of the one intake, most of which is excreted within 21 h which is 82.3% of the total urine excretion. Exclusion from feces within 6 d accounted for 21.9% of one intake, and excretion from feces within 48 h accounted for 86.2% of total excretion from feces(Table 1).
Calcium channel modulators
Mechanisms of calcium channel modulators:Calcium ion channels play an importantrole in many physiological processes of the nervous system such as the regulation of excitability of neurons, the release of transmitters at synapse, synaptic plasticity, and gene transcription. These processes are achieved by regulating of calcium ion influx.Calcium channel modulators reduce the excitement of pain conduction pathways by inhibiting the influx of calcium ions and reducing the release of neurotransmitters,thereby achieving the purpose of pain relief[12].
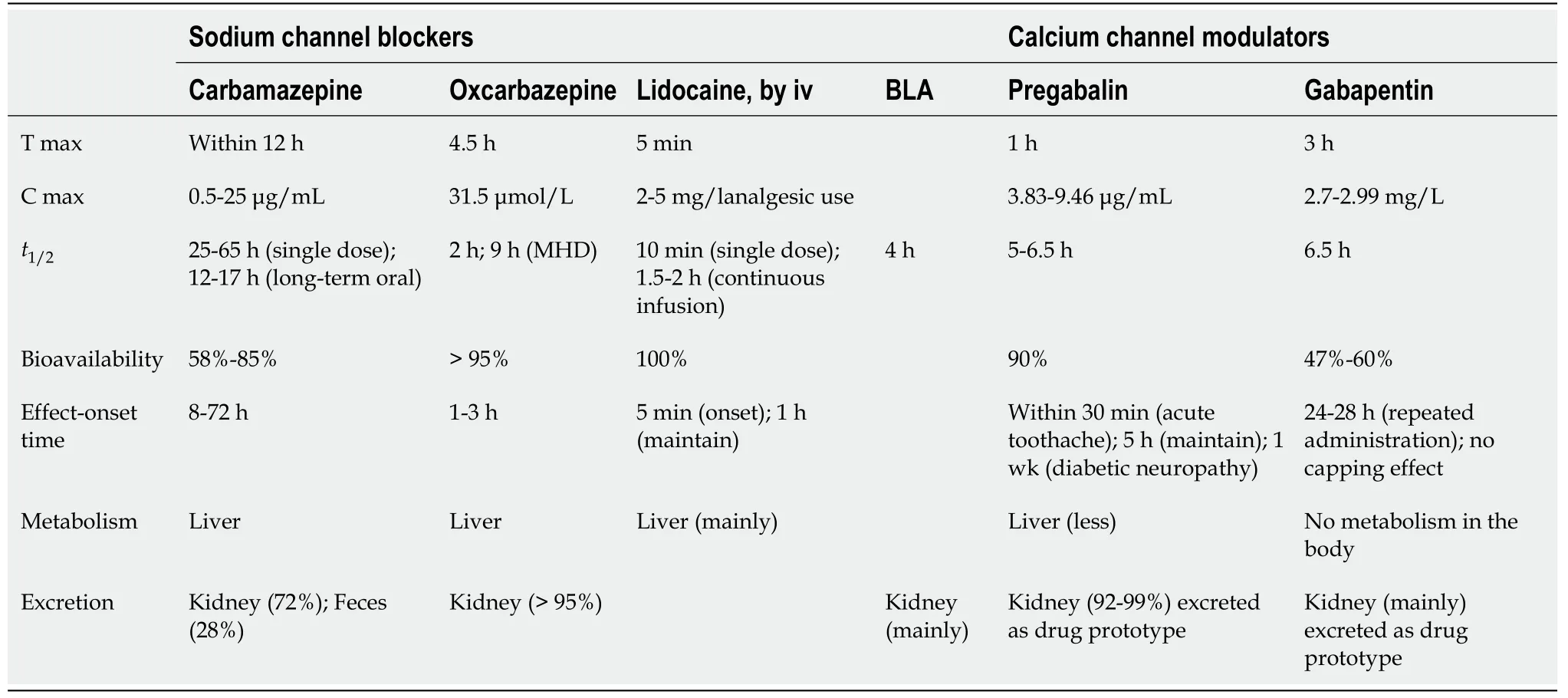
Table 1 Pharmacodynamics of ion channel drugs
Typical calcium channel modulators:Typical drugs of calcium channel modulators include gabapentin and pregabalin. Gabapentin was first used to control seizures and then was also used to treat NPP. Its structure is similar to that of GABA, but it does not work by binding with GABA receptors nor does it affect GABA synthesis and uptake.The voltage-gated calcium channel α2δ-1 subunit is the site of action of gabapentin.Specific binding can block the transport of α1 units of calcium channels from the cytoplasm to the cell membrane in dorsal root ganglia and spinal dorsal horn neurons.At the same time, the axoplasmic transport of the α2δ-1 subunit from the dorsal root ganglia to the spinal dorsal horn can also be blocked by gabapentin. Gabapentin can also inhibit the occurrence of pain through other sites such as transient receptor voltage channels, NMDA receptors, protein kinases, and inflammatory factors[13].Similar to gabapentin, pregabalin blocks extracellular calcium ion influx in order to reduce the release of excitatory amino acids.
PKs, pharmacodynamic, and physicochemical characteristics of calcium channel antagonists:Gabapentin is mainly absorbed through the active transportation of amino acid transport systems in the intestine, and a ceiling effect occurs when saturated dose is reached. Oral gabapentin has high and dose-dependent bioavailability. Oral administration 300 mg need approximate 3 h to reach peak plasma at 2.7-2.99 mg/L. It can penetrate the blood-brain barrier, and the concentration of the drug in the cerebrospinal fluid can reach 9% to 14%[14]of the dosage after oral administration. Gabapentin has a distribution volume of about 0.6 to 0.8 L/kg and a half-life of about 6.5 h. The drug does not bind to plasma proteins, is not metabolized in the body, and is excreted from the kidneys as a prototype. Its clearance is consistent with creatinine clearance rate (CCR) (Table 1).
Pregabalin is an artificial synthetics compound by amino acid and natural neurotransmitter analogue, is water- and fat-soluble as well, easily cross the bloodbrain barrier, and no GABA-like biological activity. Pregabalin is widely absorbed by the intestine after oral administration. It works within 30 min for acute toothache and sustains for 5 h. For diabetic neuropathy, onset time is about 1 wk after administration.The peak time of plasma concentration is about 1 h, and its absorption is fast and its therapeutic dose is non-linearly related to the plasma concentration. The bioavailability of pregabalin is 90%. Pregabalin is less metabolized in the liver; 92% to 99% of pregabalin is excreted by the kidney as a prototype and less than 0.1% of the oral amount is excreted with feces. The half-life is 5 to 6.5 h (Table 1).
CLINICAL APPLICATION OF ION CHANNEL DRUGS
Sodium channel antagonists
Clinical indication:Carbamazepine and oxcarbazepine are the first choice for the treatment of glossopharyngeal neuralgia and trigeminal neuralgia (TN). The International Association for the Study of Pain, Neuropathic Pain Special Interest Group and other association’s guidelines and consensus have recommended carbamazepine and oxcarbazepine as first-line treatments for primary TN.Carbamazepine has also been recommended as the first-line treatment for glossopharyngeal neuralgia[15-19]. In addition to TN, it can also be used as a second-line medication for other NPPs such as diabetic peripheral neuropathy and postherpetic neuralgia (PHN)[20](Table 2).
Lidocaine is commonly used formulation is 2% injection and 5% cream/patch. Two percent lidocaine injection is mainly used for regional nerve block treatment. It was shown that intravenous infusion of low dose lidocaine has good analgesic effect for PHN, TN, complex regional pain syndrome (CRPS), diabetic peripheral neuropathy,tumor patients with radiotherapy and chemotherapy, peripheral neuralgia, cancer pain, fibromyalgia and other NPP[21,22].
Five percent lidocaine cream or patch is used to treat acute herpes zoster neuralgia and PHN, diabetic peripheral NPP, post-traumatic NPP, sunburn, or hyperalgesia caused by capsaicin. Topical lidocaine can be used as first-line treatment for herpes zoster-related neuralgia, and the commonly used formulation is lidocaine cream and patches. The U.S. Food and Drug Administration authorized 5% lidocaine patches may be used for zoster-related NPP. For persistent pain and allodynia caused by NPP not related to herpes, it is also effective, especially for later ones[23]. It is often recommended for peripheral NPP rather than central NPP. For secondary pain caused by central nervous system damage, intravenous lidocaine (5 mg/kg) is effective for specific pain and hyperalgesia.
BLA has good anti-inflammatory, analgesic and immunomodulatory effects.Currently, tablets and capsules are commonly used. Combined with opioids, it can reduce the dose of opioids and the incidence of adverse effects. It is widely used for the treatment of rheumatic immune disease, osteoarthritis, cancer pain, and other chronic pain diseases[24].
Administration and dosage:The recommended initial dose of carbamazepine for analgesia is 100 mg, twice per day. The maintenance dose is 400-800 mg/d, several times per day. The maximum dose is 1200 mg/d. The recommended initial dose of oxcarbazepine is 150 mg, twice per day, with a maintenance dose of 300-600 mg/d and a maximum dose of 1800 mg/d. Five percent lidocaine patch/cream is used for nondamaged skin, and its patch should cover the most painful areas. Five percent lidocaine patch can be used up to three patches at one time according to the prescribed amount, and the cumulative patch time within 24 h should not exceed 12 h. Five percent lidocaine cream is applied at 1.5-2.0 g/10 cm2. Most literature recommends intravenous lidocaine at 5 mg/kg, and the infusion time should not be less than 90 min[25]. The dosage of BLA is 0.4 mg capsules or tablets, one tablet (capsule), twice to three times per day.
Side effects and precautions:(1) Carbamazepine: It has a narrow treatment window and is prone to inducing adverse effects on many systems. Common side effects are blurred vision, dizziness, fatigue, nausea, and vomiting, which mostly occur after 1-2 wk of treatment. Rash, urticaria, liver dysfunction, and hypothyroidism are seldom.Granulocytopenia and bone marrow suppression, arrhythmia, liver, and kidney failure are rare. Guidance from the Clinical Pharmacogenetics Implementation Consortium[26]states that serious diseases such as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis caused by carbamazepine are highly correlated with patients’ variant alleles human leukocyte antigen-B (HLA-B)*15:02 and HLA-A*31:01.Precaution: Carbamazepine treatment should be strictly evaluated by risk/benefit assessment, follow-up of patients with hematuria, liver and kidney function, and monitoring of plasma-concentration if possible. Attention should be paid to drug interactions with acetaminophen, digitalis, phenobarbital, erythromycin and so forth,and adverse effects should be treated in a timely manner. Attention should also be paid to hyperalgesia caused by carbamazepine withdrawal. At the same time, patients should be informed of the risk of SJS and toxic epidermal necrolysis beforeadministration. HLA-B and HLA-A genotypes can be checked when conditions are warranted. HLA-B and HLA-A genotype information will provide evidence for drug selection.

Table 2 Guidelines/expert consensus recommendations for ion channel drugs
(2) Oxcarbazepine: The most common adverse effects are rash, dizziness, headache,and drowsiness. The total incidence of adverse effects is 45.22%, most of which are mild and will alleviate or disappear after 3 to 4 wk. Oxcarbazepine does not affect liver drug enzyme metabolism and has linear PKs, and at the same time has less effects on cognitive function than barbiturates. Oxcarbazepine also can cause a severe rash, such as life-threatening SJS and toxic epidermal necrolysis, with a lower incidence than carbamazepine (1 to 6/10000vsoxcarbazepine is 0.5 to 6/1000000)[27]. Unlike carbamazepine, the correlation between oxcarbazepine’s SJS or toxic epidermal necrolysis with HLA-B1502 has not been consistently reported[28].
(3) Side effects of intravenous lidocaine: Drowsiness, paresthesia, muscle tremor,convulsions, fainting, confusion and respiratory depression, hypotension, bradycardia,and other adverse effects. Extreme high blood concentration can cause slow atrial conduction velocity, atrioventricular block, and inhibit myocardial contractility and reduce cardiac output. Precautions of intravenous lidocaine: infusion should be in an environment with monitoring and rescue conditions. During the medication, blood pressure, electrocardiogram, and other vital signs should be monitored. The concentration, total dose, and rate of infusion should be strictly controlled. Lidocaine has slow metabolism and accumulation effect in the body, hence can cause poisoning and convulsions.
Side effects of the 5% lidocaine patch are moderate skin reactions such as erythema and rash. Even with three patches per day for 12 h or even four patches for 18 h, the blood concentration of lidocaine is still very low. However, the use of lidocaine patches should be avoided in patients with oral class I antiarrhythmic drugs such as mexiletine and patients with severe liver impairment. Lidocaine gel is effective in both herpes-related neuralgia and tactile pain and is ineffective against human immunodeficiency virus neuropathy. Patients who are allergic to amide local anesthetics or other ingredients in the product are contraindicated.
(4) BLA: No drug tolerance, no addiction, no gastrointestinal adverse effects. Very few patients may experience transient mild palpitation, nausea, numbness of the lips,and palpitations. The response is transient and can be relieved after stopping the treatment. Pregnant and lactating women, children, and those allergic to this product are prohibited. The interval between the two doses should be no less than 6 h.
Calcium channel modulators
Clinical indications:Indications for calcium channel modulators are mainly NPP including peripheral NPP such as herpes zoster neuralgia, diabetic peripheral neuralgia, CRPS, central NPP such as stroke pain, and pain after spinal cord injury. It is also considered a complementary treatment for cancer pain[29]. Gabapentin is commonly used in the treatment of herpes zoster neuralgia, diabetic painful neuropathy, cancerous pain, and TN. Pregabalin has good effects on the treatment of PHN, diabetic peripheral NPP, fibromyalgia, and others (Table 2).
Administration and dosage:Gabapentin usually starts at 300 mg daily, three times a day, and needs to be titrated slowly to an effective dose. The usual dose is 900-1800 mg per day. Pregabalin is a new-generation drug developed on the basis of gabapentin.The initial dose is 150 mg daily, twice daily, and the common dose is 150-600 mg per day. To avoid dizziness and drowsiness, you should follow the principle of the first dose at night, using a small amount, gradually increasing the amount, and slowly reducing it.
Drug side effects and precautions:(1) Common adverse effects of gabapentin include dizziness, drowsiness, ataxia, and peripheral edema. Symptoms of withdrawal include disturbance of consciousness, disorientation, non-specific gastrointestinal reactions,hyperhidrosis, tremor,etc.Precautions: Because it is excreted from the kidney as a prototype, patients with renal impairment should take reduced dose. Hemodialysis can accelerate the clearance of gabapentin, so patients with hemodialysis should take increased doses to maintain an effective concentration. There is no clear evidence that gabapentin can be used in pregnant women, and it is contraindicated in lactating women because the drug can be secreted through lactation.
(2) Pregabalin adverse effects include peripheral edema, PR interval extension,dizziness, somnolence, ataxia, headache metabolism, language barriers, tremors.Endocrine adverse effects are body weight increase, the occurrence rate is 4% to 12%;the muscular skeletal adverse effect is elevated creatine kinase levels, myoclonus and case-reported stripes muscle dissolution; pregabalin treatment occasionally causes liver enzyme transient increase with mild level, saliva deficiency, constipation,thrombocytopenia, blurred vision, diplopia, amblyopia,etc.[30]. Precautions: For patients with liver damage, there is no need to adjust the dosage. The recommended dose is applied for patients with CCR ≥ 60 mL/min. In cases of patients with renal dysfunction, the dose should be adjusted. For a patient undergoing hemodialysis treatment, the dose of pregabalin should be adjusted daily according to renal function.During treatment, patients should be monitored for depression, suicidal thoughts or behaviors, and any abnormal changes in mood or behavior.
CONCLUSION
The CASP organized first-line pain management experts from China to write an expert consensus as a reference for using ion channel drugs. Here, we reviewed the mechanism and characteristics of sodium and calcium channel drugs, and developed recommendations for the therapeutic principles and clinical practice for carbamazepine, oxcarbazepine, lidocaine, BLA, Pregabalin, and gabapentin. We hope this guideline provides direction for clinicians and patients when they use ion channel drugs for the management of NPP.
 World Journal of Clinical Cases2021年9期
World Journal of Clinical Cases2021年9期
- World Journal of Clinical Cases的其它文章
- Expert panel’s guideline on cervicogenic headache: The Chinese Association for the Study of Pain recommendation
- Expert consensus of Chinese Association for the Study of Pain on the application of ozone therapy in pain medicine
- Chinese Association for the Study of Pain: Experts consensus on ultrasound-guided injections for the treatment of spinal pain in China(2020 edition)
- Chinese Association for the Study of Pain: Expert consensus on diagnosis and treatment for lumbar disc herniation
- Expert consensus on the diagnosis and treatment of myofascial pain syndrome
- Chinese Association for the Study of Pain: Expert consensus on chronic postsurgical pain