
2型糖尿病的代谢免疫
2021-04-01 01:12尤玉青王妍之李伟周东浩
现代免疫学 2021年2期
尤玉青,王妍之,李伟,周东浩
(1.徐州医科大学 内分泌科,徐州 221004;2.滨州医学院 内分泌科,滨州 264033;3.徐州医科大学附属医院 内分泌科,徐州 221004;4.徐州医科大学附属临沂市人民医院 营养科,临沂 276000)
随着人们生活水平的提高,2型糖尿病(type 2 diabetes mellitus, T2DM)在全球范围内广泛流行。T2DM是以胰岛素抵抗为主伴有β细胞功能障碍及血糖水平升高为特征的代谢紊乱性疾病。近年来,代谢和免疫之间复杂而密切的联系逐渐被人们所重视,在此基础上诞生了一门崭新的学科——代谢免疫学[1]。T2DM作为典型的代谢性疾病,同样也存在着复杂而深刻的代谢和免疫之间的相互作用,在不同的组织涉及到不同的信号通路转导过程[2]。那么,饮食诱导的肥胖是如何在机体不同组织中诱发炎症进而影响糖代谢以及胰岛素抵抗的?这个问题引起了越来越多人的关注。
1 白色脂肪组织
白色脂肪组织不仅包含脂肪细胞,还包含前脂肪细胞、成纤维细胞、内皮细胞以及免疫细胞等在内的基质细胞。脂肪细胞除了可以储存脂肪外,还能起到内分泌与旁分泌的作用,通过分泌瘦素、TNF-α、IL-6以及脂联素等多种生物活性物质引起局部和全身炎症反应,最终导致代谢紊乱[3]。
一方面,长期高糖高脂饮食会通过脂肪细胞继发性肥大和增生,来满足储存更多脂质的要求,但当脂肪细胞的体积增至一定范围时,会因血管生成相对不足导致缺氧。细胞缺氧可激活低氧诱导因子1α(hypoxia inducible factor 1α,HIF-1α),而HIF-1α则通过抑制血管生成因子的表达进一步加重缺氧,最终导致细胞凋亡。当死亡细胞增至一定量时,可诱导免疫细胞积聚,促使炎症反应的发生。大脂肪细胞可分泌单核细胞趋化因子1(monocyte chemoattractant protein 1,MCP-1)、游离脂肪酸(free fatty acid,FFA)以及瘦素等物质。MCP-1可募集循环单核细胞至脂肪组织并分化成巨噬细胞,同时可诱导巨噬细胞从M2型极化为M1型;而瘦素则通过促进内皮细胞的激活来募集巨噬细胞。M1型巨噬细胞可分泌TNF-α、IL-6等促炎因子来诱导炎症,同时也可通过促炎因子来激活核转录因子-κB-IκB激酶(nuclear factor-κ-light-chain-enhancer of activated B cells-inhibitor ofκB kinases,NF-κB-IKK)与c-Jun N-末端激酶-激活蛋白1(c-Jun-N-terminal kinases-activator protein 1,JNK-AP-1)等炎症通路形成级联反应,导致炎症加剧。另一方面,来自肠-肝轴的代谢性内毒素中的LPS与FFA会通过与TLR4结合来诱导NF-κB-IKK以及丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)途径的活化,最终导致大量促炎因子的产生[4-6]。
脂肪组织中的正常胰岛素信号转导涉及胰岛素与脂肪细胞上的胰岛素受体结合,诱导胰岛素受体底物1(insulin receptor substrate 1,IRS-1)蛋白磷酸化、磷脂酰肌醇3激酶(phosphatidylinositol-3-kinase,PI3-K)活化、蛋白激酶B(protein kinase B,Akt)活化以及雷帕霉素靶蛋白活化等。一方面,促炎因子如TNF-α会通过降低细胞膜上胰岛素浓度调节葡萄糖转运蛋白4(glucose transporter 4,GLUT4)的活性来降低胰岛素依赖性葡萄糖的摄取;另一方面,炎症通路如NF-κB-IKK与JNK-AP-1会分别通过IKKβ与JNK抑制IRS-1磷酸化来阻断下游胰岛素信号的转导。因脂肪细胞摄取葡萄糖减少,导致外周胰岛素释放量增加,最终造成胰岛素敏感性的降低。另外,慢性炎症以及HIF-1α可促使间质发生纤维化来降低细胞外基质(extracellular matrix,ECM)弹性,从而造成脂肪组织纤维化,导致异位脂质积聚[5,7-9]。
2 肝脏
肝脏由肝细胞、肝免疫细胞以及肝星状细胞等构成。源自脂肪组织的FFA经血液循环可在肝细胞内蓄积,并且与内毒素共同造成肝细胞损伤;受损的肝细胞会释放IL-1α激活肝脏的固有免疫细胞(Kupffer细胞)。Kupffer细胞通过分泌MCP-1诱导单核细胞来源的巨噬细胞的募集;活化的免疫细胞会分泌一系列的细胞因子如TNF-α、IL-6、白三烯等,并且与炎症信号通路共同加剧炎症反应。过度的炎症反应通过破坏肝细胞脂代谢造成肝脏脂肪变性,同时还增加神经酰胺的产生。神经酰胺能通过激活蛋白磷酸酶2A破坏Akt/PKB胰岛素转导信号通路,导致胰岛素抵抗[10]。
肝脏中的炎症不仅会将肝星状细胞转变为肌成纤维细胞,还会激活成纤维细胞。肌成纤维细胞与成纤维细胞可同时合成并分泌大量的ECM,使ECM大量沉积于狄氏间隙内,造成肝细胞生长因子的抑制以及胶原酶的合成与分泌,最终导致肝脏纤维化。肝脏纤维化致使正常糖代谢的肝细胞急剧减少,造成外周高血糖并引起相应的代偿性高胰岛素血症,最终导致胰岛素敏感性降低[11]。
3 骨骼肌
肥胖会引起肌细胞间脂肪组织(intermyocellular adipose tissue,IMAT)和肌肉周围脂肪组织(perimuscular adipose tissue,PMAT)扩张,从而导致巨噬细胞、T细胞等免疫细胞渗入2种脂肪组织中;同时免疫细胞会极化成促炎表型,致使IMAT和PMAT发生炎症反应;这2者的炎症会导致肌细胞分泌并释放促炎因子、趋化因子以及血管紧张素Ⅱ(angiotensin Ⅱ,ANG-Ⅱ)等大量生物活性物质,从而加剧肌细胞的炎症反应。
细胞因子不但可以通过直接激活炎症途径使IRS-1磷酸化,还可以通过产生INF-γ激活Janus激酶/信号转导和转录激活因子(Janus kinase/signal transducer and activator of transcription,JAK/STAT)途径中断胰岛素信号转导,导致胰岛素抵抗。同时炎症也可降低肌细胞膜上的GLUT4的表达,导致肌细胞减少葡萄糖的摄取。ANG-Ⅱ可通过减少骨骼肌的血供、刺激活性氧的产生以及激活炎症途径等多种方式直接破坏肌细胞的胰岛素转导,最终导致胰岛素抵抗[12]。
4 胰腺
胰腺包括外分泌腺与内分泌腺。外分泌胰腺含有腺泡细胞,而内分泌胰腺则分泌如胰岛素、胰高血糖素等各种激素。肥胖可促使外分泌胰腺内脂肪浸润,造成FFA的大量释放从而损伤腺泡细胞;受损的腺泡细胞则会通过激活NF-κB等转录因子释放大量的细胞因子如IL-1β以及其他炎性介质。这些介质通过向胰腺募集大量中性粒细胞、巨噬细胞等免疫细胞加剧炎症反应,炎症可反过来促使腺泡细胞大量死亡以及激活胰蛋白酶原造成大量胰酶的释放[13-14]。一方面,炎症和胰酶共同作用于内分泌胰腺,导致胰岛素分泌减少以及胰高血糖素分泌增加,两者共同导致机体内血糖升高。另一方面,细胞因子如IL-1β可与β细胞膜上的受体结合,不仅可通过细胞因子信号转导抑制因子1的活化和IRS-2/Akt信号转导的破坏,导致胰岛素信号转导障碍,还可以直接诱导β细胞凋亡,减少胰岛素的分泌[14-15]。
同时,为了应对外周胰岛素抵抗,胰岛通常通过增加胰岛素分泌来维持机体血糖,这一过程称为β细胞补偿。这是通过扩大胰岛β细胞体积,增加胰岛素生物合成以及通过增强对葡萄糖、FFA和胰高血糖素样肽1刺激的敏感性来完成的。但随着时间的推移,胰岛β细胞逐渐衰竭,无法完成这一补偿,最终导致高血糖[16]。
5 中枢神经系统
为了响应外周代谢和免疫信号的变化,中枢神经系统中的几种细胞类型也随之发生变化。FFA可活化小胶质细胞,活化的小胶质细胞可释放促炎因子,导致神经炎症的发生;炎症则会进一步活化周细胞,活化的周细胞与星形胶质细胞共同导致血脑屏障的破坏,进而使其渗透性增加。外周循环中细胞因子、脂肪酸以及瘦素等物质进入到大脑中,加剧神经炎症[17]。
下丘脑可以调节食物摄入、能量消耗以及葡萄糖代谢。细胞因子以及炎症信号通路会破坏瘦素以及胰岛素信号转导;瘦素与胰岛素共享转导信号通路,而且胰岛素还增强下丘脑中瘦蛋白诱导的信号转导级联反应;瘦素可以减少食物摄入并增加能量消耗。同时,神经炎症会激活与食物摄入、能量消耗以及葡萄糖代谢相关的下丘脑神经元回路。瘦素与胰岛素的信号转导以及下丘脑神经元回路的异常最终会导致瘦素抵抗以及胰岛素抵抗[18]。
6 胃肠道
长期高糖高脂饮食可导致肠道微生物失调,进而影响微生物相关产物以及内源性代谢物的变化。一方面,宿主与肠道微生物的共生作用维持肠道免疫系统的稳定,因而微生物的变化会相应引起嗜酸性粒细胞以及巨噬细胞等肠道固有层免疫细胞群的变化,导致肠道炎症以及肠道通透性的改变。另一方面,微生物代谢产物如短链脂肪酸会促进肠上皮细胞的增殖与分化,维持肠黏膜完整性,因而短链脂肪酸的减少会导致肠黏膜通透性增加;而肠道屏障的破坏,则进一步导致肠道内细菌进入血液循环,形成代谢性内毒血症,并且革兰阴性杆菌的细胞壁成分LPS可通过激活TLR4诱导胰岛素靶细胞中的炎症反应[19-21]。
肠道内源性代谢物如胆汁酸会通过激活回肠和肝脏中的核法尼醇X受体以及肠内分泌的胆汁酸G蛋白偶联受体5,产生餐后信号,因而胆汁酸的减少会导致机体不断摄入食物[22]。 (图1)
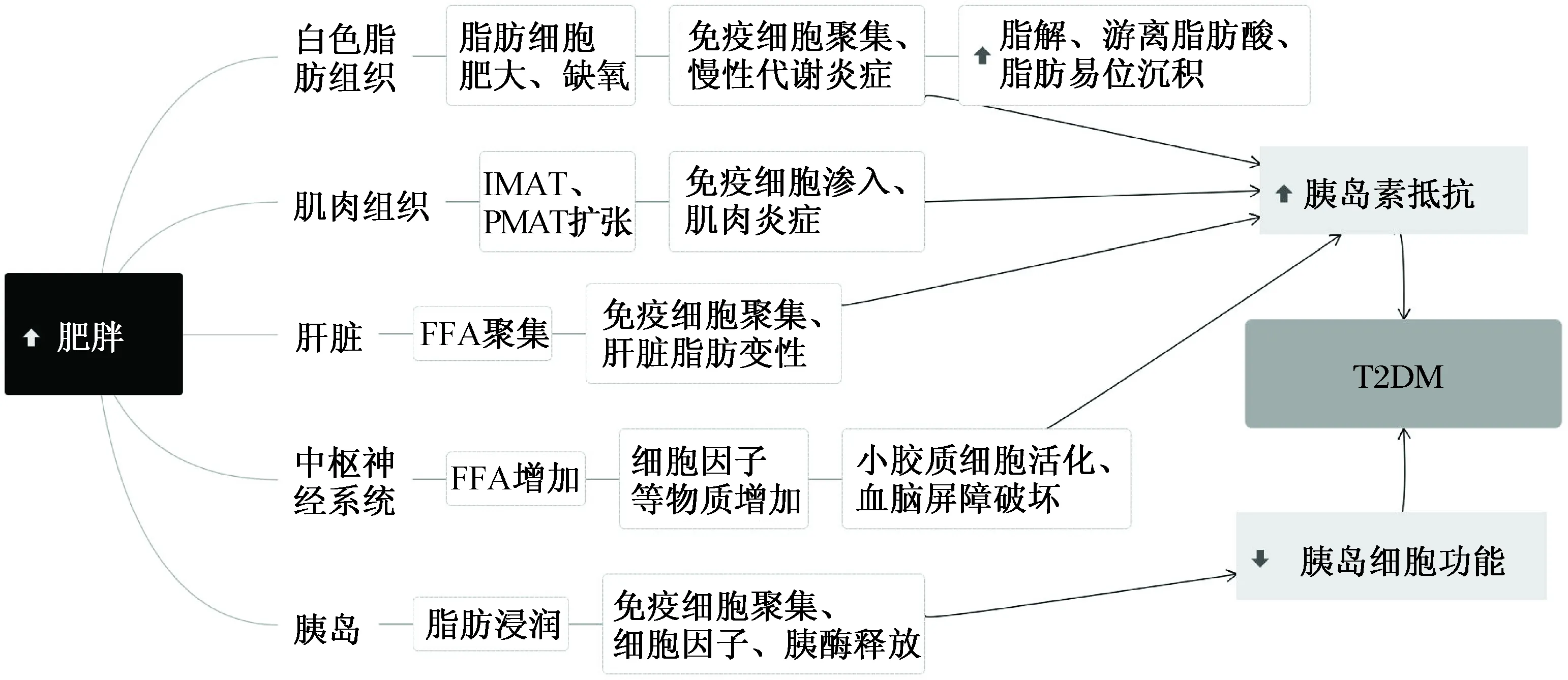
图1 T2DM起始不同组织的代谢免疫变化
7 治疗
7.1 饮食对于DM的饮食控制,不仅包含食物的热量,任何导致肠道微生物失衡以及通透性增加的因素,都应考虑在内。已有大量研究证据表明,食品添加剂如乳化剂及甜味剂可改变肠道微生物,导致肠道紊乱和炎症,促进代谢综合征的发展。因此,应尽量减少食品添加剂[23]。
同时含有大量饱和脂肪酸的饮食会通过各种途径影响胰岛素敏感性以及糖耐量,应减少摄入富含饱和脂肪酸的食物,如乳制品和肉类等动物源性食物以及椰子和棕榈油等一些蔬菜源性食物;同时,应多食用富含不饱和脂肪酸的食物如鱼油,改善饮食诱导的炎症,以此来改善胰岛素抵抗[9]。
7.2 药物原则上来说,任何可以减少机体炎症的药物都可以改善糖耐量以及胰岛素敏感性。一方面,因为T2DM患者的β细胞中IL-1β表达增加,这会促进β细胞凋亡,所以可以通过抗IL-1β抗体抑制免疫细胞因子的作用,治疗T2DM,这已在人类中进行了研究,结果表明可适度改善血糖。此外已有研究证实,使用白三烯B4受体1抑制剂治疗的肥胖小鼠,3种胰岛素靶组织(肌肉、肝脏和脂肪)对胰岛素的敏感性更高,糖耐量改善[24-26]。
另一方面,可以通过抑制炎症通路来阻断炎症级联反应,目前研究最多的是水杨酸盐及其衍生物。已有一项小型临床研究表明,大剂量水杨酸盐可能通过抑制FFA引起的IRS-l丝氨酸磷酸化,发挥改善胰岛素抵抗的作用[27]。
此外,G蛋白偶联受体120(G protein-coupled receptor 120,GPR120)可能成为DM的潜在治疗靶点。已有研究显示,缺乏GPR120的小鼠在喂食高脂饮食时会导致葡萄糖代谢受损和脂肪生成增加。GPR120不仅可通过破坏TLR4与TNF-α促炎信号通路发挥抗炎作用,而且可以抑制下丘脑中小神经胶质细胞的活化。ω-3多不饱和脂肪酸也可通过GPR120来阻断炎症信号转导途径,降低巨噬细胞趋化性和抑制巨噬细胞表型的转变,从而改善胰岛素敏感性。因此,高亲和力的小分子GPR120激动剂可抑制炎症反应和胰岛素抵抗以及改善葡萄糖耐量[17,28]。
8 结语
在脂肪、肝脏、胰腺及中枢神经系统等不同组织中,T2DM都存在着复杂而深刻的代谢与免疫之间的相互作用。目前,虽然抗炎疗法已经在DM治疗方案中占据一席之地,但任何事物都具有两面性,正如化疗药物治疗恶性疾病时,并不会区分细胞的好坏一样,抗炎方法也可能对免疫系统的某些方面产生不必要的抑制,给机体带来不利影响。因此,使用抗炎疗法治疗代谢性疾病,应仔细评估,权衡利弊,使抗炎方案趋利避害,最终开启DM治疗的新篇章。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
昆明医科大学学报(2021年3期)2021-07-22
云南医药(2021年3期)2021-07-21
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
长春中医药大学学报(2017年1期)2017-04-16
现代检验医学杂志(2016年4期)2016-11-15
中国民族医药杂志(2016年6期)2016-05-09
吉林大学学报(医学版)(2015年1期)2015-12-17
中国医药导报(2015年24期)2015-02-28
