
基于Keap1-Nrf2-A RE信号通路对多发性硬化症治疗的研究及展望
2021-04-01 06:28:42刘国栋庄春林余建强
宁夏医科大学学报 2021年2期
刘国栋,庄春林,余建强,石 英
(1.宁夏医科大学药学院,银川 750004;2.第二军医大学药学院,上海 200433)
多发性硬化症(multiple sclerosis,MS)是一种中枢神经系统(central nervous system,CNS)退行性疾病,会导致大脑和脊髓白质中的局灶性炎症性脱髓鞘,从而产生运动和认知功能障碍[1-2]。有证据表明MS的炎性反应与氧化应激密切相关,氧化应激反应通过氧化脂质、蛋白质和DNA等直接促进脱髓鞘,也间接地通过诱导免疫失调和炎性反应而导致脱髓鞘[3-4]。Keap1-Nrf2-抗氧化反应元件(antioxidant response element,ARE)信号通路是一个综合的氧化还原反应体系,激活该途径对于维持细胞的氧化平衡至关重要。该通路在炎性反应中也起着关键作用,因此,清晰认识Keap1-Nrf2-ARE信号通路各部结构和功能及整体作用机制在MS研究过程中有着重要意义。
1 Keap1-Nrf2-ARE信号通路的结构
1.1 Nrf2
Nrf2是具有高度保守性的碱性亮氨酸拉链结构的转录因子(图1)。人类的Nrf2由605个氨基酸组成,含有7个结构功能域,分别被命名为Neh1~Neh7,每个结构域对Nrf2的功能起着不同的作用[5]。Neh1是一个包含碱性亮氨酸拉链基序,可与细胞核内DNA、小肌腱膜纤维肉瘤蛋白(Maf)或其他转录因子形成异二聚体,从而再与ARE结合,启动目标基因的转录。Neh2位于Nrf2的N端,含有多个可被泛素化修饰的赖氨酸残基和两个Keap1的结合位点(ETGE和DLG),这两个基序对于Keap1和Nrf2之间调节Nrf2泛素化和稳定性的相互作用至关重要[6]。Neh3位于C末端,通过与Nrf2转录辅助激活子染色质解螺旋酶DNA结合蛋白6结合活化转录。Neh4和Neh5是两个独立的转录结构域,当Nrf2进入细胞核并以Nrf2-Maf二聚形式与ARE结合后,并不能立即启动转录,还需要cAMP反应元件结合蛋白与Neh4、Neh5两个结构域结合,启动转录过程。Neh6是一个富含丝氨酸残基的结构域,对于Nrf2有独立于Keap1的负性调控作用[7]。Wang等[8]在研究视黄醇受体α(RXRα)与Nrf2相互作用的时候发现Nrf2中未定义的氨基酸209-316区域,将其命名为Neh7结构域。
1.2 Keap1
Keap1是由624个氨基酸组成的蛋白质,负调控Nrf2的活性。人类的Keap1蛋白序列包含5个结构域,分别为N端区域(N-terminal region,NTR)、BTB区域(broad complex,tramtrack,and Bric-aBrac,BTB)、中间连接区域(linker intervening region,IVR)、DGR区域(double glycine repeat,DGR)及C端区域(C-terminal region,CTR),见图1[9]。人类Keap1蛋白共包含27个半胱氨酸残基(Cys),其中7个Cys(Cys151、Cys257、Cys273、Cys288、Cys297、Cys434和Cys613)对活性氧和亲电试剂具有较高的活性,参与氧化还原信号传导[6,10]。BTB结构域的主要功能是促进Keap1与Nrf2的二聚和Keap1与Cul3的相互作用。IVR结构域具有对氧化物或亲电试剂高度敏感性的半胱氨酸残基,可作为氧化应激的传感器。DGR结构域包含6个双甘氨酸重复序列,是Keap1与Nrf2的Neh2区连接的蛋白接触位点。DGR和CTR域也被称为DC结构域,它们与Nrf2的Neh2结合,介导Keap1和Nrf2之间的相互作用[6-7]。
1.3 ARE
ARE也被称为亲电效应元件,位于许多细胞保护基因启动子的上游,调节具有细胞保护作用蛋白和Ⅱ相代谢酶基因的表达。在氧化应激的条件下,游离的Nrf2转位进入细胞核,与小肌腱膜纤维肉瘤蛋白结合形成异二聚体,激活依赖ARE目的基因的表达,发挥抗氧化作用。研究表明,Nrf2与Bach1均可以与ARE结合,启动Ⅱ相代谢酶和抗氧化基因的表达,从而保护机体组织细胞的正常功能[11-12]。
1.4 Keap1-Nrf2-ARE信号通路的活化
在正常的生理状态下,Keap1通过其BTB结构域形成一个同二聚体,然后与细胞质中Nrf2的DC结构域结合,通过泛素化-蛋白酶体途径使Nrf2降解。而当机体受到亲电试剂刺激或处于氧化应激状态时,Keap1的半胱氨酸残基被共价修饰,导致其构象发生改变,与Nrf2解离,从而避免了Nrf2被泛素化和降解。累积的Nrf2进入细胞核与ARE结合,从而启动下游一系列保护基因的表达,如NADPH醌氧化还原酶-1(NADPH:quinone oxidoreductase-1,NQO-1)、血红素加氧酶-1(Heme oxygenase-1,HO-1)、谷胱甘肽-S-转移酶(glutathione-S-transferase,GST)等,见图2。研究显示,Nrf2信号通路调控600多个基因,其中200多个编码与炎症、神经退行性疾病和其他慢性疾病相关的蛋白质[13]。
2 Nrf2激活剂
激活Nrf2的方式主要有共价和非共价修饰两类。共价修饰是通过配体亲电性基团与Keap1中的硫醇等随机反应结合,其反应性比较难控制,与目标靶点的结合缺乏特异性,从而降低其调控作用,最终导致一些不必要的脱靶毒性效应。非共价修饰是通过直接阻断Keap1与Nrf2之间的相互作用,进而使Nrf2从复合物中解离,发挥激活Nrf2的作用。非共价修饰克服了共价Nrf2激活剂的脱靶问题,避免了不确定的毒副作用[10,14]。两者作用机制见图3。
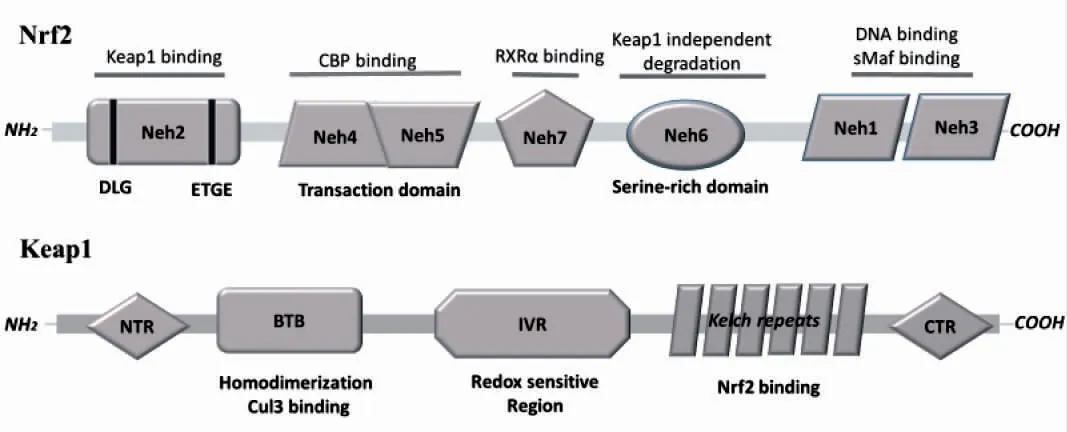
图1 Keap1和Nrf2结构区域
2.1 Nrf2亲电激活剂
由图4可见,富马酸二甲酯(Dimethyl Fumarate,DMF)为化合物1,是一个成功的Nrf2激活剂,已被FDA批准用于治疗复发型多发性硬化症[14]。药代动力学研究表明,DMF是一种在体内迅速转化为富马酸单乙酯的前药,与Keap1的Cys151残基共价反应,导致Nrf2介导的脑神经元和神经胶质细胞基因反式激活。然而DMF的临床应用会产生一些不良反应,如轻度至中度腹痛、腹泻、恶心和严重白细胞减少等症状[15]。巴多索隆(Barduxolone,CDDO)是另一个Nrf2亲电激活剂,它与Keap1的Cys151残基发生共价可逆反应。但CDDO甲基化修饰的产物——甲基巴多索隆(Barduxolone methyl,CDDO-Me)为化合物2,因增加了糖尿病患者心力衰竭的风险,未能通过肾脏治疗的Ⅲ期临床试验[16]。CDDO-Me目前正在肺动脉高压疾病的临床试验中进行研究[15]。萝卜硫素为化合物3,是西兰花芽中提取的异硫氰酸酯化合物,在一些临床研究中也得到了评价[14,17]。
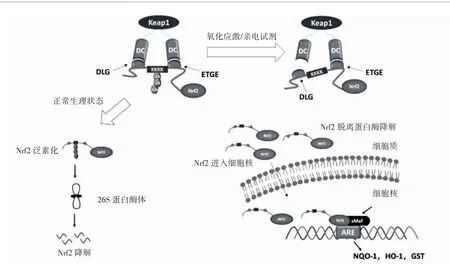
图2 Keap1-Nrf2-ARE信号通路活化机制

图3 不同Nrf2激活剂的作用机制

图4 Nrf2亲电激活剂
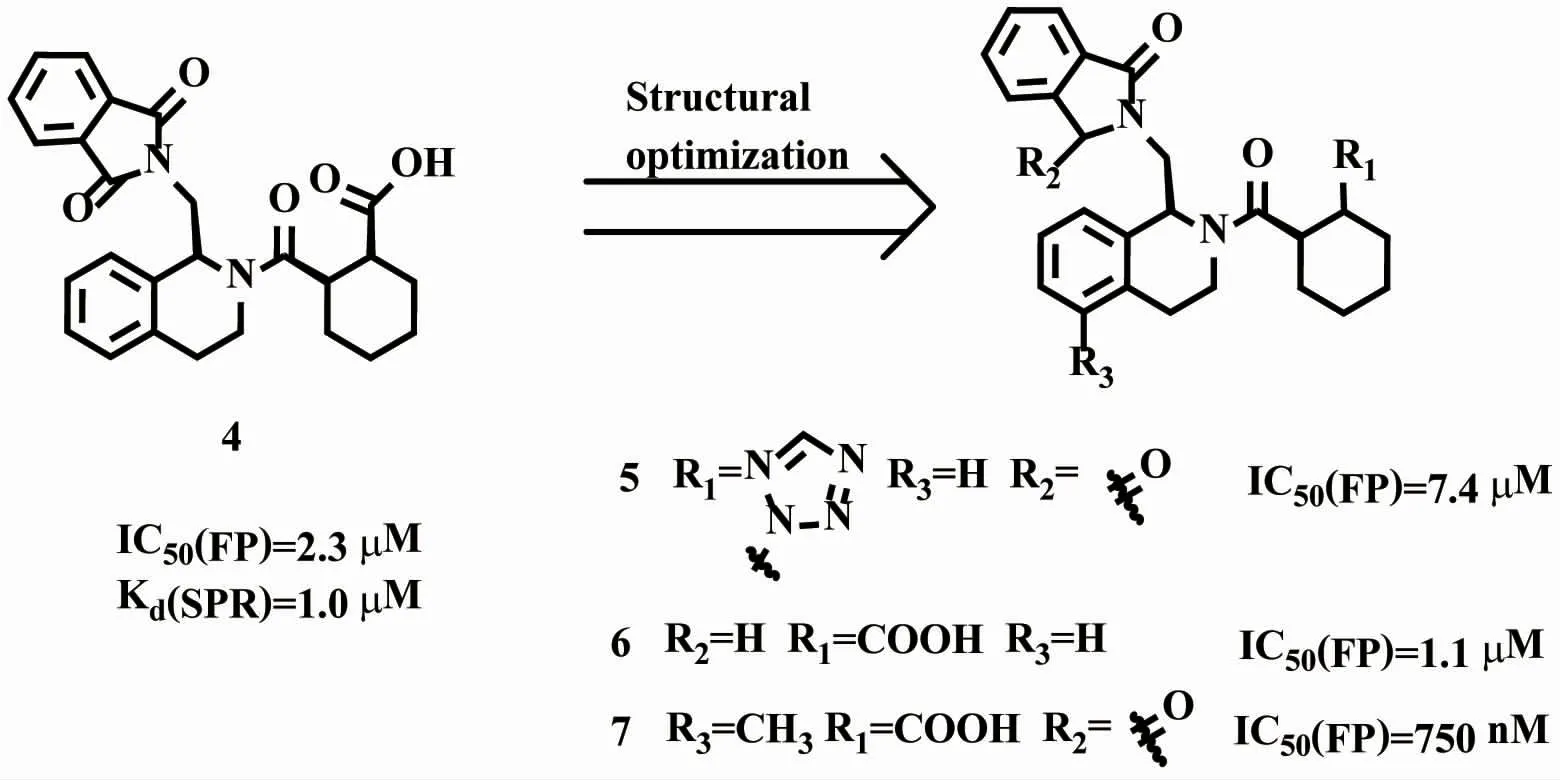
图5 四氢异喹啉类结构优化
2.2 Nrf2非共价激活剂
在已开发非共价激活剂的设计和优化过程中运用了一些经典的药物化学策略,如基于结构的药物设计和优化策略、基于生物电子等排原理、基于片段的药物设计策略和基于骨架跃迁的药物优化策略等,从而使目标化合物靶点亲和力和蛋白-蛋白相互作用抑制活性普遍得到提升,同时使理化性质更有助于化合物成药。同时,进一步通过有效地激活Nrf2对于MS的研究具有广阔的应用前景,加快了MS治疗药物的研发进程。
2.2.1 基于结构的药物设计和优化策略 Hu等[18]首次报道了1,2,3,4-四氢异喹啉类的小分子Keap1-Nrf2抑制剂(图5)。图5中化合物4通过高通量筛选得到,通过荧光偏振(fluorescence polarization,FP)测定法测定其对Keap1-Kelch结构域与Nrf2衍生肽之间相互作用的抑制活性(IC50=2.3μM)。通过表面等离子体共振(Surface plasmon resonance,SPR)测得其对靶点的亲和力值Kd=1.0μM。然后在化合物4的基础上,利用生物电子等排体原理将羧酸用四氮唑取代,得到化合物5,其对Keap1-Nrf2抑制活性降低了约3倍(IC50=7.4μM);然而当去除邻苯二甲酰亚胺的羰基得到化合物6,其对Keap1-Nrf2抑制活性提高约2倍(IC50=1.1μM);在5位引入甲基得到化合物7,其对蛋白相互作用抑制活性进一步提升(IC50=750 nM)[19]。
2.2.2 基于生物电子等排的优化策略 Marcotte等[20]通过筛选267551个化合物得到以1,4二氨基萘为母核的Keap1-Nrf2抑制剂化合物8,其对蛋白相互作用的抑制活性为IC50=1.46μM;Jiang等[21]在化合物8的基础上进行了进一步结构优化,在化合物8的磺酰胺基的氮上引入亚甲基羧酸得到化合物9。化合物9是更有效的Keap1-Nrf2抑制剂,相对于化合物8抑制活性提高了约200倍以上(IC50=28.6 nM),并且靶点亲和力也大大提高(Kd=3.59 nM);为了改善化合物9的类药性,利用对乙酰氨基取代对甲氧基得到化合物10,其对蛋白相互作用的抑制活性进一步提升(IC50=14.4 nM)[22];Lu等[23]利用生物电子等排原理将羧酸用四氮唑取代,得到化合物11,使其在保持原抑制活性的基础上透膜性大大提升(Pe=42.266±4.476),见图6。
2.2.3 基于片段的药物设计策略 2016年,Astex制药公司和葛兰素史克制药公司通过基于片段的药物设计方法报道了一种新型苯丙酸类Keap1-Nrf2抑制剂[24]。通过利用X射线晶体衍射筛选出大约330个片段,鉴定出分别与关键氨基酸残基Arg483、Tyr525和Ser602相互作用的A、B、C片段,尽管片段A对蛋白相互作用抑制活性>1 mM,但仍被确定为细化的“锚片段”。在片段生长过程中,苯丙三唑部分直接连接到片段A的苄位,从而得到了化合物12(IC50=61μM)(图7)。在化合物12氯苯的3位引入烷基苯磺酰胺,由于苯磺酰胺可以与Tyr334通过π-π堆积相互作用,进而使活性进一步提升,得到化合物13(IC50=0.27μM),见图7。最后,为了使目标分子能够以稳定构象与靶点结合,通过苯磺酰胺的环化反应,形成了一个融合的7元杂环,得到活性进一步提升的化合物14(图7)[6,16]。研究表明,化合物14可以激活慢性阻塞性肺病(chronic obstructive pulmonary disease,COPD)患者支气管上皮细胞中的Nrf2通路,从而诱导靶基因表达,提高抗氧化活性,减轻炎性反应。同时发现,化合物14可以激活COPD大鼠模型中的Nrf2,使得肺组织中炎症程度降低[24]。
2.2.4 基于骨架跃迁的结构优化策略 由图8可见,化合物15为已报道的非共价Keap1-Nrf2抑制剂,其蛋白相互作用抑制活性为0.11μM(图8)。但是萘环1、4位上两个氮原子会引起代谢稳定性问题[25]。Abed等基于骨架跃迁策略将化合物15的萘环减化为一个苯环,得到1,2-二取代二甲苯类似物16[26],其代谢稳定性虽然增加,但抑制活性降低。通过在化合物16羧酸α碳上引入甲基,得到了抑制活性提高约15倍的手性化合物17(IC50=0.15μM)。同样化合物19也是基于骨架跃迁策略减化化合物18中一个苯环得到的4-苯基-1,2,4三唑母核Keap1-Nrf2抑制剂,化合物19的硝基通过与关键氨基酸残基Arg483相互作用,有效激活Nrf2-ARE通路,同时其对靶点亲和力也进一步提升(Kd=16.5μM)[16]。
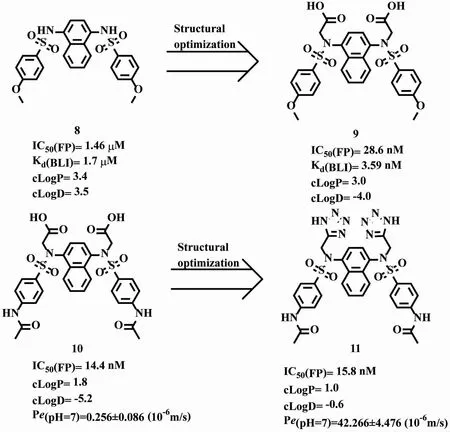
图6 1,4-二氨基萘类结构优化
3 MS治疗药物现状
MS的药物治疗始于20世纪90年代,干扰素β和醋酸格拉替雷是1993年批准的第一批治疗MS注射制剂,两者主要用于治疗复发缓解型多发性硬化症。10年后,第一种单克隆抗体那他珠单抗获得批准,它是一种人源化的单克隆抗体,可防止淋巴细胞穿过血脑屏障[27]。芬戈莫德,特立氟胺和富马酸二甲酯是在2010至2020年上市的三个口服药物,其中芬戈莫德于2018年5月被FDA批准用于治疗10~25岁的青少年患者[28-30]。研究表明,自20世纪90年代以来,全球MS患病率急剧上升。目前,全球有250多万的MS患者,其中80%被确诊为复发缓解型,这相比于1990年患病率增加了10.4%[2,31]。国内外首推的MS黄金治疗方法是疾病修饰治疗(disease-modifying treatments,DMT)。同时根据中国《多发性硬化患者生存报告(2019)》可知,国内多发性硬化症患者3万~5万人,83%为复发缓解型,临床上接受DMT治疗的仅有10%,仍有90%的患者因为副作用和间断性用药导致复发等各种原因而处于无药可用的状态,因此,急需一种新的治疗手段和策略。

图7 苯丙酸类的药物设计
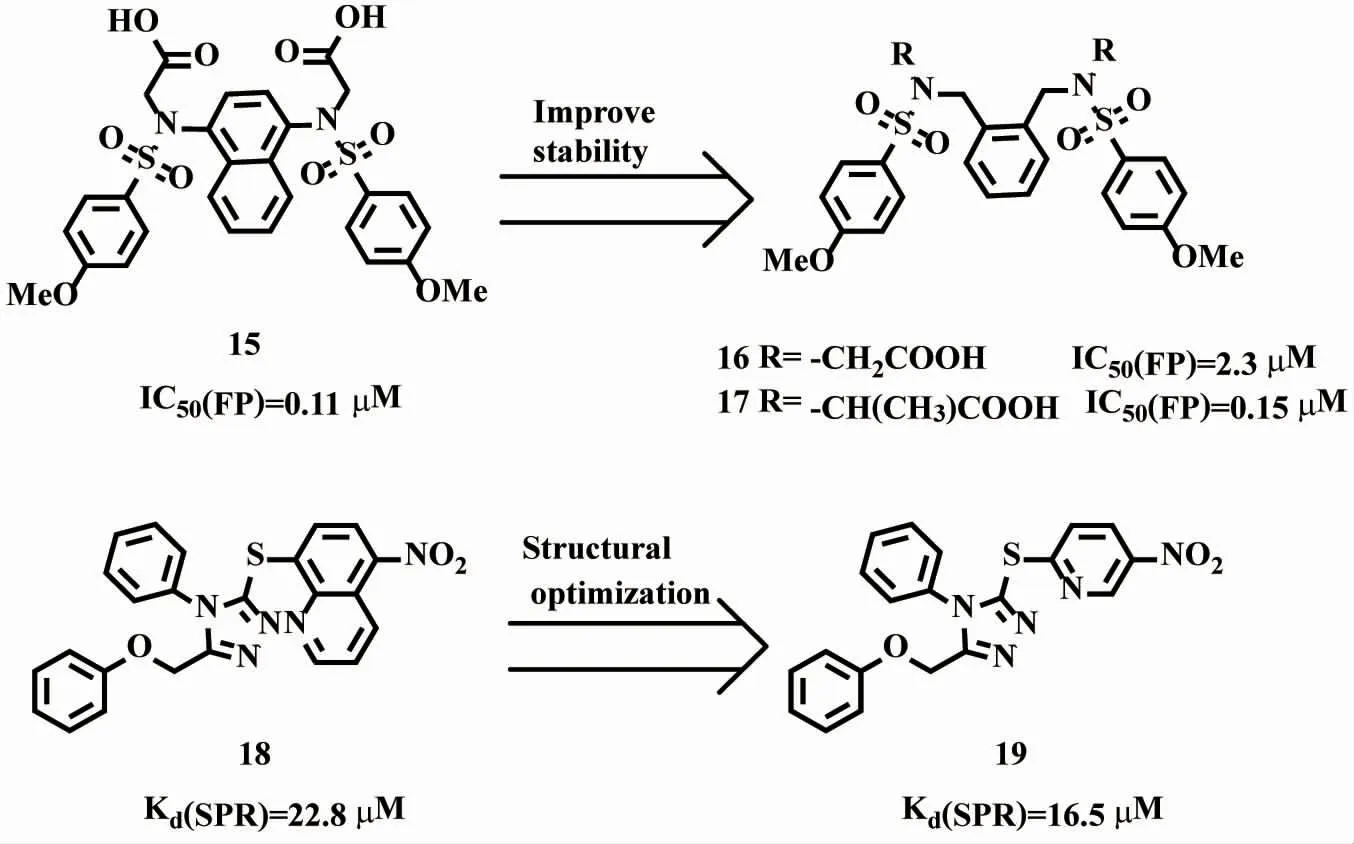
图8 1,2-二取代二甲苯类似物结构设计
4 激活Nrf2能够有效缓解MS
目前,MS仍不能被彻底治愈,急需寻找和开发新的治疗途径。相关研究表明,激活Nrf2能够调控免疫炎性反应、铁代谢、线粒体功能、自噬等方面,进而缓解MS患者和动物模型的临床症状。
4.1 CNS的炎症调节
在经典MS模型——实验性自身免疫性脑脊髓炎(experimentally induced autoimmune encephalomyelitis,EAE)模型中研究发现,DMF作为Nrf2激活剂,可显著缓解脱髓鞘和认知功能障碍等临床症状[32-33]。在MS的病理过程研究中发现,高促炎性自身反应T细胞入侵到中枢神经系统,产生干扰素γ和白细胞介素17A导致炎性反应[4]。同时,增加了血脑屏障的通透性,并促使其他免疫细胞进入中枢神经系统[4,34]。该过程伴随着巨噬细胞/小胶质细胞的持续激活,并释放促炎细胞因子和趋化因子,使炎性反应加剧[35-36]。研究发现,在MS患者和EAE动物模型中,Nrf2低表达且HO-1缺乏时,导致EAE小鼠模型脱髓鞘加剧和患者病情恶化[37-38]。而当激活Nrf2时,下游白细胞介素17以及炎症因子Th1和Th17的产生受到抑制,同时使下游HO-1高表达,从而抑制促炎细胞因子和趋化因子的过度分泌,最终缓解炎性反应[39-40]。
4.2 铁代谢
脑内铁代谢失调是MS疾病过程中导致氧化应激的重要因素之一[41-42]。在健康的成人脑中,铁主要储存在少突胶质细胞中。而在MS患者脑中的巨噬细胞和小胶质细胞铁含量也是增加的,这些过量的铁在髓鞘降解和清除的过程中会被释放到细胞外,随后转化为二价亚铁形式,通过Fenton反应生成活性氧进一步对机体造成损伤[43]。另外,巨噬细胞中铁的含量过高也会促使炎症的发生[44]。在EAE模型中,脑中铁过量会导致参与磷脂和髓鞘代谢的几个相关蛋白表达降低,可能会影响髓鞘的合成和恢复[45]。激活Nrf2可以调节铁代谢中的铁蛋白及相关酶来维持体内铁平衡,降低脱髓鞘程度,进而缓解MS的临床症状和疾病复发[46]。
4.3 线粒体功能
越来越多的证据表明,线粒体功能障碍也是MS发病和复发的重要因素之一[47-48]。通过对MS患者大脑轴突研究发现,线粒体功能障碍和ATP缺乏,会导致动作电位在传导过程中无法将Na+从轴浆转移到细胞外[49-50],这会导致反向Na+-Ca2+交换体的激活,造成轴浆内Ca2+浓度升高,最终引起Ca2+介导的中枢神经系统炎症,并伴随能量失衡和脱髓鞘[51-52]。通过对分离线粒体和培养细胞的研究表明,Nrf2缺乏可导致线粒体脂肪酸氧化、呼吸和ATP生成等过程受阻[53-54]。同时研究发现MS患者中过氧化物酶体增殖物激活受体γ-辅助活化因子-1α(peroxisome proliferatoractivated receptor gamma coactivator 1 alpha,PGC-1α)的表达降低会导致线粒体功能障碍,从而促进复发,加快病情进展[55-56]。Nrf2也被报道通过激活PGC-1α和核呼吸因子1来维持线粒体正常的能量代谢功能[57]。因此,通过激活Nrf2来维持MS患者体内线粒体功能稳态,对于缓解MS相关临床症状具有十分重要的意义。
4.4 自噬
研究显示,自噬降解途径受阻会导致MS细胞外纤维连接蛋白的聚集,进而影响再髓鞘化过程[58]。MS患者由于自噬水平的降低,在大脑和脑脊液中均检测到可溶性低聚物以及在EAE模型中也检测到氧化蛋白聚集体增加,进而加剧炎性反应[59-61]。研究表明,在EAE模型中,Nrf2缺乏导致自噬降解途径降低,而通过激活Nrf2可诱导自噬相关基因的表达,进而促进自噬,缓解脱髓鞘和炎性反应[62]。
非共价小分子Keap1-Nrf2抑制剂由于分子量较大,且含有大极性亲水性基团难以透过血脑屏障进入中枢神经。为了克服这种局限性,可以通过生物电子等排等结构优化策略,使目标分子保持原活性的基础上具有更强的血脑屏障透过能力。此外,在既患有癌症又患有MS的患者治疗过程中,Nrf2激活剂会增加肿瘤转移的风险,这也取决于疾病的背景和阶段[63,64]。因此,在使用Nrf2激活剂药物治疗该类患者时,要评估此类药物的风险。
5 总结和展望
MS是一种发病机制复杂的多因素疾病,目前研究的药物虽尚不能彻底治愈,但通过调控Keap1-Nrf2-ARE信号通路,能够有效缓解MS的复发和临床症状。其中激活Nrf2能够使MS患者及动物模型中的免疫炎性反应、铁代谢、自噬等生理过程介导的临床症状得到改善。大多数Nrf2亲电激活剂由于反应性比较难控制,与目标靶点的结合缺乏特异性,最终导致一些不必要的脱靶毒性效应。而非共价小分子Nrf2激活剂通过阻断Keap1和Nrf2之间的相互作用克服了亲电性Nrf2激活剂的缺点,非共价Nrf2激活剂在MS的研究中具有良好的应用前景,未来很有可能会用于MS的临床治疗。同时,结合国内的MS治疗情况,增加对Keap1-Nrf2-ARE信号通路在MS过程中所起作用的认识,有助于新型MS治疗药物的研发。
猜你喜欢
现代临床医学(2023年4期)2023-09-26 09:41:24
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
潍坊学院学报(2021年2期)2021-07-22 07:59:12
高等学校化学学报(2021年7期)2021-07-11 16:25:48
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:34:54
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:36
中国卫生标准管理(2015年6期)2016-01-14 05:17:18
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
