
磁性MnO2-Fe3O4复合氧化物催化5-羟甲基糠醛氧化合成2,5-呋喃二甲酸研究
2021-03-27 09:24卢虹竹白继峰张新月王景芸周明东
燃料化学学报 2021年3期
卢虹竹,白继峰,闫 飞,张新月,金 颖,王景芸,*,陈 平,周明东,*
(1.辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001;2.辽宁省产品质量监督检验院,辽宁 沈阳 110032)
5-羟甲基糠醛(HMF)可由果糖、葡萄糖和纤维素降解脱水得到[1,2],是一种重要的生物基平台化合物,经还原、加氢、氧化和聚合可转化为多种高附加值化学品和生物燃料[3,4],尤其是HMF氧化可生成 2,5-二甲酰基呋喃(DFF)、5-羟甲基-2-呋喃甲酸(HMFCA)、5-甲酰基-2-呋喃甲酸和 2,5-呋喃二甲酸(FDCA)[4,5]。在这些呋喃类化学品中,FDCA与对苯二甲酸(PTA)具有相似的化学结构,可以替代PTA合成聚2,5-呋喃二甲酸-乙二醇酯、聚2,5-呋喃二甲酸-1,4-丁二醇酯等聚酯,也可以替代异酞酸、己二酸、琥珀酸等合成聚酯、聚酰胺、环氧树脂等生物基聚合物,被美国能源部称为12个最有前途的生物基平台化合物之一[6]。
铬酸盐、重铬酸盐和高锰酸盐等强氧化剂均可以用于氧化HMF氧化反应,但此类氧化剂有毒,且用量较大[7]。均相催化体系Co/Mn/Br对HMF氧化反应具有一定的促进作用,但该催化体系不仅难以与反应体系分离,还会导致严重的设备腐蚀和环境污染[8,9]。因此,将非均相催化剂应用于HMF氧化反应逐渐引起人们的关注。Au、Pt、Pd和Ru等贵金属在高压氧气条件下对HMF氧化反应具有良好的催化作用[10]。Davis等[11]比较了Pt/C、Pd/C、Au/C和Au/TiO2等不同负载型贵金属催化剂对HMF氧化反应的催化效果和产物分布。结果表明,此类催化剂对HMF氧化反应均表现出相当的催化活性,在690 kPa O2压力条件下,催化剂Au/C和Au/TiO2可以获得较高收率的中间氧化产物HMFCA,而Pt/C、Pd/C可以将HMF氧化为最终氧化产物FDCA。Ait等[12]也以Pt/C为催化剂进行 HMF氧化反应,在 373 K和 40 bar O2压力条件下可获得70% FDCA收率。尽管贵金属基催化剂对HMF氧化制备FDCA的反应表现出良好的催化活性,但由于催化剂成本过高,催化活性不稳定,此类催化剂在HMF氧化反应中的应用受限。因此,将非均相廉价过渡金属基催化剂用于促进HMF 氧化反应具有重要的实际意义。Mn[13-16]、Fe[17-19]、Co[20,21]等 金 属 的 氧 化 物 和 Mo[22,23]、 Fe[24,25]、Co[24,26]、Cu[27]等金属的配合物对HMF氧化反应均有一定的催化能力[28],在这些非贵金属基催化剂中,金属氧化物由于制备方法简单、合成成本较低而引起了广泛关注。MnO2是催化HMF氧化反应的有效催化剂,但使用单一的MnO2氧化物难以高选择性地得到FDCA[29,30],通常采用添加第二种金属氧化物的方法提高催化剂的活性和选择性。Mn基金属氧化物虽然在HMF氧化反应中表现出很好的催化性能,但氧化剂多为高压氧气或空气,反应操作不易控制[16,29,30]。此外,为提高FDCA选择性,反应多在碱性水溶液中进行,需要大量酸中和反应液,产生大量工业废水[14,15,21]。因此,开发廉价、绿色的催化剂在温和条件下进行HMF氧化反应制备FDCA具有重要的意义。磁性Fe3O4不仅对氧化反应表现出较好的催化活性,且由于具有磁性易于分离循环使用,因此,除可单独作为催化剂使用外,常与MnOx、CeO2等金属氧化物复配并用于HMF氧化反应[18,20]。结果表明,Fe3O4的引入能够显著提高催化剂的活性和选择性。不同晶型的MnO2催化性能不同,本研究比较不同晶型MnO2对HMF氧化反应的催化作用,并引入Fe3O4形成复合金属氧化物MnO2/Fe3O4,并对MnO2-Fe3O4催化的HMF氧化反应行为进行研究。
1 实验部分
1.1 实验试剂与催化剂的制备
试剂:五水合硫酸锰、四水合乙酸锰、过硫酸铵、三氯化铁六水合物、硫酸亚铁七水合物、尿素(99%,500 g,上海麦克林生化科技有限公司),聚乙二醇(98%,500 g,天津天泰精细化学品有限公司),无水乙醇(98%,500 mL,天津市恒兴化学试剂制造有限公司,),高锰酸钾、氢氧化钠(98%,500 g,国药集团化学试剂有限公司),5-羟甲基糠醛(98%,25 g,Aladdin),5-羟甲基-2-呋喃羧酸、2,5-呋喃二甲醛、5-甲酰-2-呋喃甲酸、2,5-呋喃二甲酸(98%,1 g,Aladdin)。
不同晶型的MnO2与Mn-Fe复合氧化物的制备:参照文献方法[16,20,31]合成不同晶型的MnO2与Mn-Fe复合氧化物,并根据Mn、Fe原子比例不同将复合氧化合物表示为 MnzFeyOx(z/y= 8/3、3/8、1/1)。
磁性 Fe3O4的制备:称取 5.40 g FeCl3·6H2O 和3.60 g 尿素溶解于 200 mL 去离子水中,90 °C 下剧烈搅拌2 h,将混合物冷却至室温,搅拌下加入2.80 g FeSO4·7H2O,滴加 0.1 mol/L NaOH 溶液,调节 pH值约为10,将浆液在室温下超声处理30 min,老化5 h,抽滤并用去离子水和无水乙醇反复洗涤,100 °C下干燥12 h,制得Fe3O4磁性氧化物。
1.2 催化剂的表征
采用德国Bruker公司D8 Advance型 X射线衍射仪(XRD)对样品进行物相分析。实验条件:Cu靶,Kα辐射源,石墨单色器,工作电压 40 kV,管电流 80 mA,10°−80°扫描,扫描速率 8(°)/min,步长为0.1°。采用日本Hitachi公司生产的SU8010型扫描电子显微镜(SEM)对样品的整体形貌进行分析,工作电压:20 kV,工作距离:15 mm,分辨率:1.5 nm;采用美国 Thermo scientific公司生产的配备铝/镁双阳极靶的PHI-5300/ESCA型X射线光电子能谱仪(XPS)对样品元素组成、价态及相对含量进行分析,能量分辨率为0.8 eV;采用美国康塔公司生产的带热导检测器的 ChemBET Pulsar TPR/TPD型自动化学吸附分析仪进行CO2和NH3吸附-脱附分析。样品于 He 气氛中(30 mol/min)在 300 ℃预热1 h后冷却至50 ℃,吸附二氧化碳和氨气至平衡,切换He吹扫10 min,最后在He气流中以10 ℃/min 的速率从 50 ℃ 升温至 850 ℃,进行程序升温脱附。采用美国Agilent公司生产的Cary 660型傅里叶变换红外光谱仪表征催化剂红外酸度,以吡啶为探针分子,表征催化剂Brönsted酸性位和Lewis酸性位。称取15 mg样品压成半透明状薄片,在红外吸收池中于300 ℃高真空净化1 h,降至室温后吸附吡啶至平衡,然后升温脱附吡啶,测定B酸和L酸。
1.3 HMF的氧化和产物分析
将 HMF (75 mg,0.6 mmol)和 70% 的叔丁基过氧化氢加入到DMSO中,搅拌至溶解,加入催化剂,在一定温度下进行反应。反应结束,利用氧化物和搅拌子的磁性将催化剂分离,然后向反应液中加入蒸馏水将其稀释,过滤后定容至100 mL,采用 Agilent 1100高效液相色谱对氧化产物进行定量分析。本论文所用液相色谱检测器为紫外检测器,测定波长为278 nm。色谱柱为C18反相色谱柱 (200 mm×4.6 mm),控制柱温为 35 ℃,以乙腈和质量分数 0.1% 的乙酸水溶液(体积比 = 30∶70)为流动相,控制流量1.0 mL/min。通过标准曲线计算反应生成的FDCA、DFF、HMFCA和FFCA等氧化产物收率,计算公式如下:

式中,wp——产物收率,%;mp、mHMF——产物和原料HMF的质量,g;MP、MHMF——产物和原料HMF的相对分子质量。
2 结果与讨论
2.1 不同晶型MnO2对HMF氧化反应的催化作用
采用水热合成法,通过调变Mn前驱体的浓度、氧化剂的种类和浓度以及反应温度,制备出三种不同晶型的MnO2氧化物,XRD分析结果见图1。图1表明,制备的三种MnO2氧化物晶型分别为α-、γ-和δ-MnO2。将得到的MnO2用于催化HMF氧化反应,比较不同晶型MnO2的催化活性。将HMF(0.6 mmol, 75 mg)、 6.5 equiv.TBHP 和 MnO2加 入DMSO溶剂中,催化剂用量为0.002 g/mL,混合均匀后在80 ℃反应12 h,结果见表1。当氧化反应在不加催化剂的条件下进行时,HMF转化率仅为40.5%,中间氧化产物HMFCA、DFF和FFCA的产率分别为4.1%、10.2%和7.8%,而并未得到最终氧化产物FDCA。加入MnO2催化剂后,HMF转化率均显著提高,表明三种晶型的MnO2对HMF氧化反应均存在一定的催化活性,尤其是α-MnO2催化的HMF氧化反应,转化率达83.2%,呋喃类氧化产物总收率达62.3%,其中,FDCA收率为13.3%。而γ-MnO2和δ-MnO2催化的HMF氧化反应呋喃类氧化产物总收率较低,虽然得到了>20%的FFCA收率,但FDCA收率均不及5.0%,表明γ-MnO2和δ-MnO2对HMF氧化为FDCA的反应催化能力不及α-MnO2。采用XPS对不同晶型MnO2中元素化学态进行分析,结果见图 2。图 2(a)为 Mn 2p的XPS谱图,结合能为641.7和653.35 eV的峰分别归属于 Mn 2p3/2和 Mn 2p1/2,Mn 2p3/2的核心峰可分裂成结合能为641和642 eV的两个峰,分别归属于 Mn3+和 Mn4+。拟合计算结果表明,α-MnO2中Mn4+相对 含 量为 54.1%, 而 γ-MnO2和 δ-MnO2中Mn4+相对含量分别为 49.3%和 49.2%。对 O 1s的信号峰进行分峰拟合,由图 2(b)可以看出,O 1s的核心峰也分裂为两个峰,结合能为529−530 eV的峰归属于表面晶格氧 O2−(标记为 Oα),结合能为531.1−532.3 eV的峰归属于氧化缺陷和表面低配位氧负离子((标记为 Oβ),而晶格氧已被证明可以显著加速氧化反应[31]。α-MnO2中Oα相对含量相对含量为59.2%,而 γ-MnO2和 δ-MnO2中 Oα相对含量均低于α-MnO2,分别为 56.8%和58.1%。由此可以看出,α-MnO2中Mn4+和表面晶格氧相对含量均高于γ-MnO2和δ-MnO2,从而使α-MnO2中催化活性中心 Mn4+·−O2−离子对数量相对较高,这是 α-MnO2催化活性高于 γ-MnO2和 δ-MnO2的重要原因。
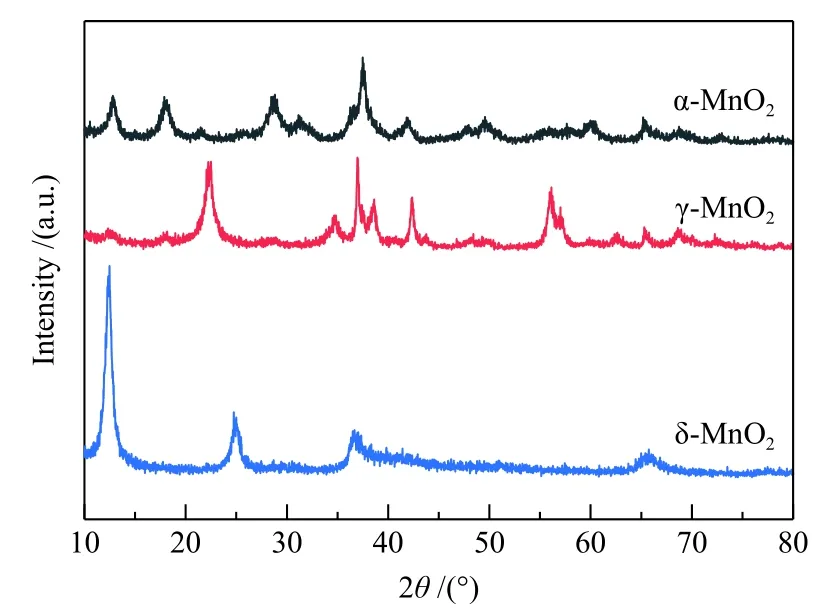
图1 不同晶型 MnO2 的 XRD 谱图Figure 1 XRD patterns of MnO2 with various crystal structures
同样,将Fe3O4用于催化HMF氧化反应,结果表明Fe3O4对HMF也有一定的催化作用,可获得76.8%的转化率,中间氧化产物FFCA收率为30.1%,但反应并未获得理想的FDCA收率,表明其对FFCA的选择性高于α-MnO2,因此,该反应体系中,单一的Fe3O4和MnO2催化剂均不能将HMF有效氧化生成FDCA,因此,本研究选择催化活性较强的α-MnO2与Fe3O4复合制备复合金属氧化物,并分析其对HMF氧化反应的催化作用。
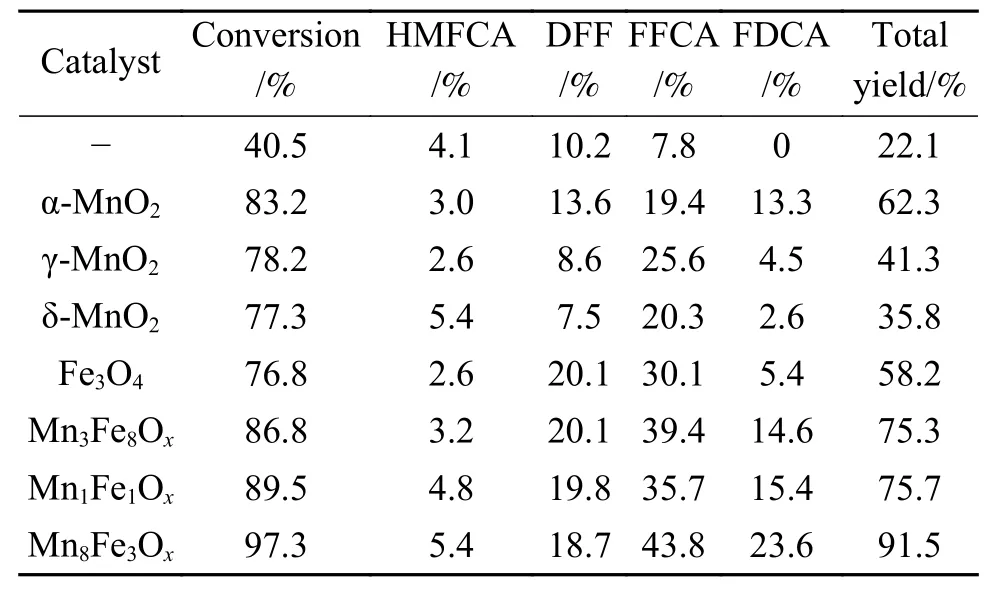
表1 不同氧化物催化的HMF氧化反应Table 1 Perfomance of different catalysts on oxidation of HMF
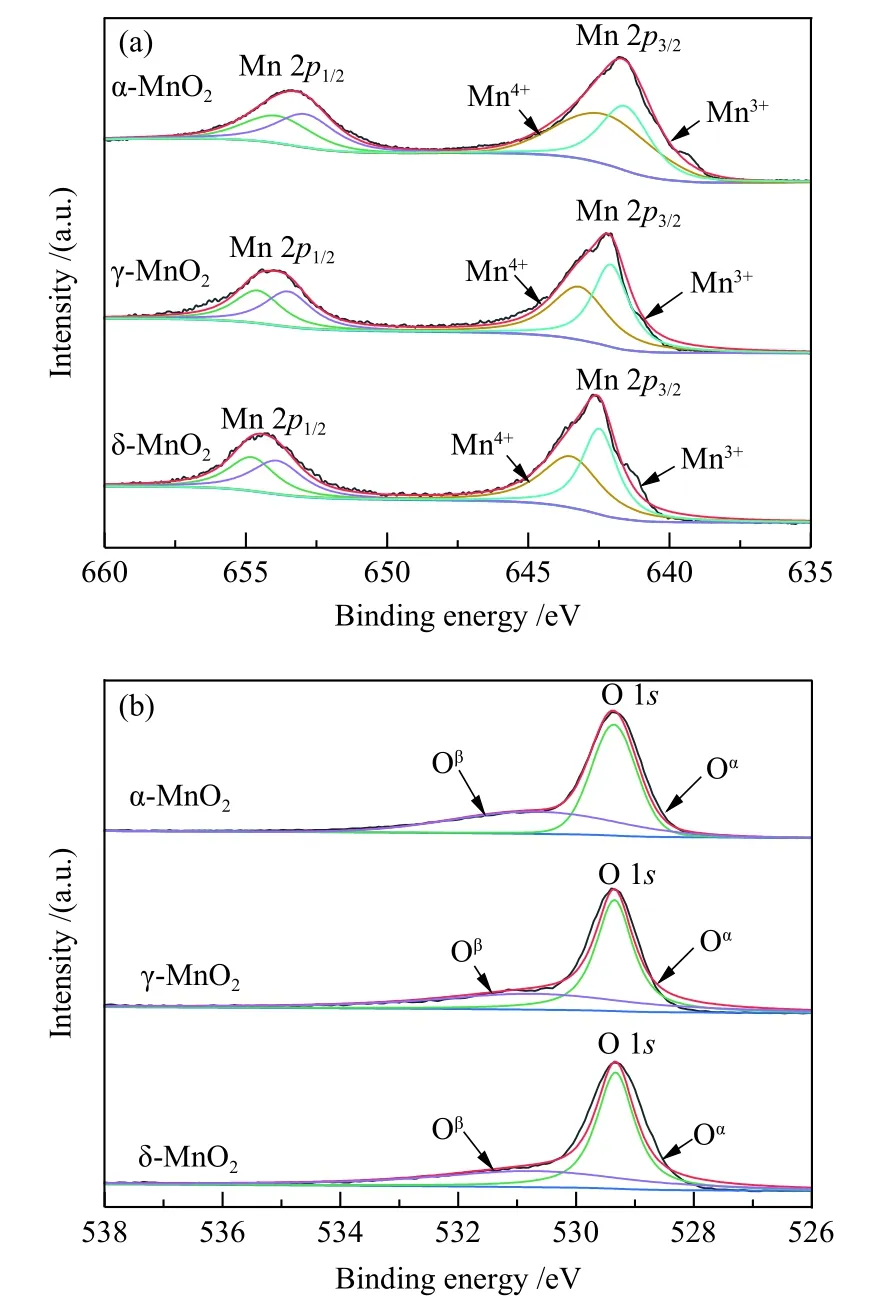
图2 不同晶型 MnO2 XPS 谱图Figure 2 XPS spectra of MnO2 with various crystal structures(a): Mn 2p; (b): O 1s
2.2 复合金属氧化物的表征
2.2.1 XRD分析
制备不同Mn/Fe原子比的复合金属氧化物(记作MnyFezOx)并进行XRD分析,结果如图3所示。Fe3O4的衍射峰出现在 30.2°、 35.6°、43.3°、53.8°、57.3°和 63.0°处,与 Fe3O4尖晶石结构一致(JCPDS No.19-0629)。由不同 Mn/Fe 原子比的复合金属氧化物XRD谱图分析可知,Fe3O4的引入使α-MnO2衍射峰强度减弱,当Mn/Fe原子比为3∶8和1∶1时,催化剂XRD谱图中未出现明显的MnO2衍射峰,说明此时未形成完整的晶核,而当Mn/Fe原子比为 8∶3时,出现 α-MnO2衍射峰,催化剂记作Mn8Fe3Ox。
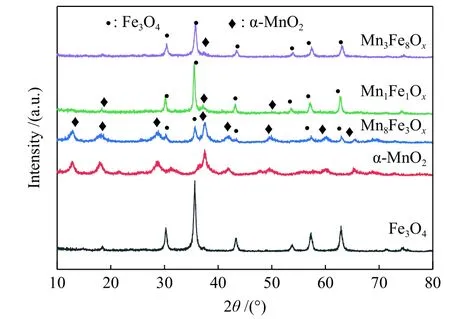
图3 Mn-Fe 复合金属氧化物的 XRD 谱图Figure 3 XRD patterns of Mn-Fe mixed oxide catalysts
2.2.2 SEM分析
Fe3O4、α-MnO2和 Mn8Fe3Ox的 SEM 照片如图4所示。由SEM照片可以看出,Fe3O4的形貌为聚集的不规则纳米粒子(a),而α-MnO2呈现直径约30 nm、长度为 100−300 nm 的纳米棒状结构(b)。形成Mn-Fe复合金属氧化物后,Fe3O4和α-MnO2基本保持原有的形貌,与单一的α-MnO2相比,Mn8Fe3Ox中α-MnO2纳米棒更加尖锐,而Fe3O4纳米粒子并未发生变化。
上述研究结果表明,形成复合金属氧化物对α-MnO2晶核的形成产生了一定的影响,但并未影响Fe3O4和α-MnO2形貌。
2.3 复合金属氧化物Mn8Fe3Ox对HMF氧化反应的催化作用
2.3.1 Mn-Fe复合金属氧化物的催化作用
以不同Mn/Fe原子比的复合金属氧化物为催化剂进行HMF氧化反应,由表1可以看出,Fe3O4的引入使复合催化剂的催化活性显著增强,转化率和各氧化产物的收率均有所增加。但XRD分析结果表明,复合氧化物中Mn/Fe原子比小于1时,未完全形成α-MnO2晶核,因此,尽管中间氧化产物FFCA收率显著增大,但FDCA收率增幅较小。Mn/Fe原子比增大至8∶3时,催化剂活性进一步增强,HMF转化率增大至97.3%,呋喃类氧化产物总产率高达91.5%,但FDCA收率仅为23.6%,仍获得了43.8%的中间产物FFCA,该结果表明,此条件下中间产物DFF和FFCA仍不能完全氧化转化为FDCA。继续增大复合氧化物中金属Mn比例,在相同条件下以Mn10Fe1Ox为催化剂进行HMF氧化反应,HMF转化率和各氧化产物的产率反而小幅度降低,表明Mn8Fe3Ox是该体系HMF氧化制备FDCA的最佳催化剂,未得到理想FDCA收率的原因可能是由于反应温度过低、反应时间过短或催化剂和氧化剂用量较少所导致。接下来分析Mn-Fe复合氧化物产生催化活性的原因。
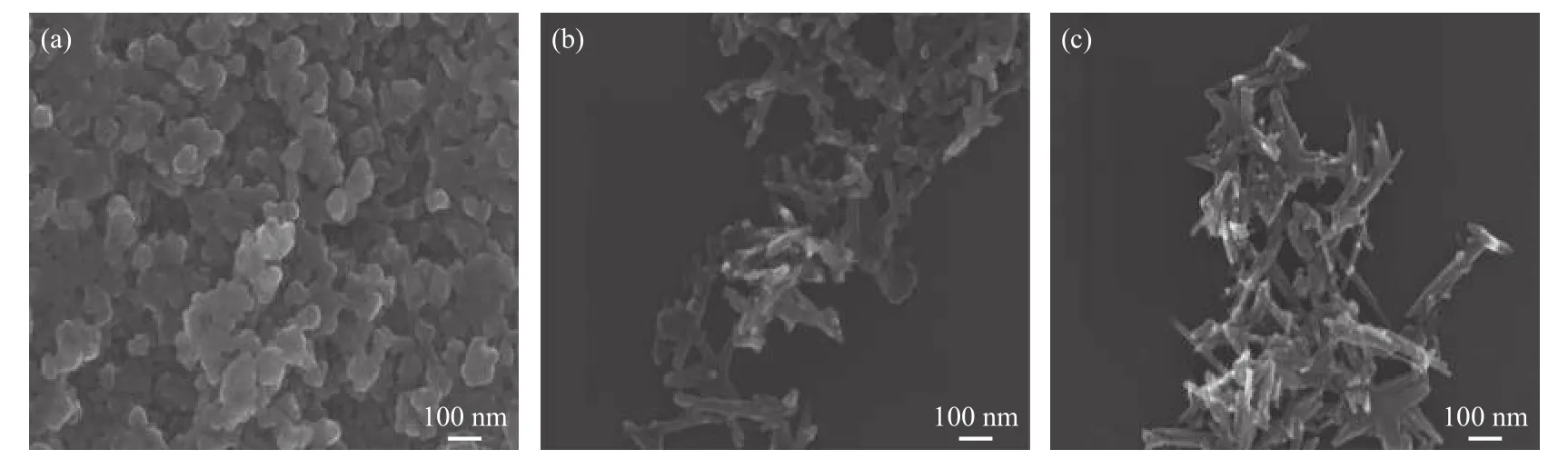
图4 Fe3O4、α-MnO2和 Mn8Fe3Ox的 SEM 照片Figure 4 SEM images of Fe3O4 (a), MnO2 (b) and Mn8Fe3Ox (c)
图5 为 MnO2、Fe3O4和 Mn8Fe3Ox的 XPS 谱图。
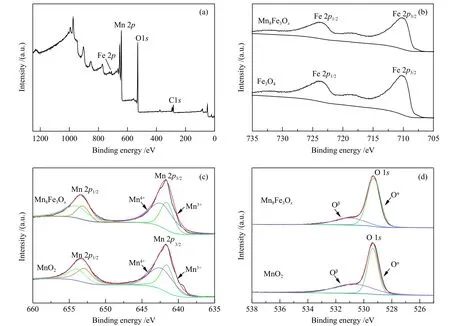
图5 Mn8Fe3Ox 和 α-MnO2 XPS 谱图:(a)Mn8Fe3Ox XPS 全扫描谱图,(b)Fe 2p XPS 谱图,(c)Mn 2p XPS 谱图,(d)O 1s XPS 谱图Figure 5 XPS spectra of Mn8Fe3Ox and α-MnO2: (a) Wide survey XPS spectrum of Mn8Fe3Ox, high resolution XPS spectra(b) Fe2p, (c) Mn2p, and (d) O 1s of Mn8Fe3Ox and MnO2
Mn8Fe3Ox的全扫描谱图(图 5(a))给出了 Mn 2p、Fe 2p和 O 1s物种的信号峰,表明催化剂 Mn8Fe3Ox由Mn、Fe和O三种元素组成。图5(b)为催化剂Fe3O4和 Mn8Fe3Ox的 Fe 2pXPS 谱 图, 结 合 能 为710.3 和 723.8 eV 的峰归属于 Fe 2p3/2和 Fe 2p1/2,是磁性 Fe3O4中 Fe2+的特征峰[32]。对 Mn 2p的信号峰进行拟合分峰,结果如图5(c)所示。拟合计算结果表明,α-MnO2中 Mn4+相对含量为 54.1%,而与Fe3O4复合后,Mn8Fe3Ox催化剂中Mn4+相对含量增加至64.7%。对O 1s的信号峰进行分峰拟合,由图5(d)可以看出,复合金属氧化物Mn8Fe3Ox中Oα相对含量由59.2%增加至75.0%。XPS分析结果表明,Fe3O4的引入使催化剂Mn4+和表面晶格氧O2−相对含量增大,从而使催化活性中心 Mn4+·O2−离子对数量增加,因此,复合催化剂Mn8Fe3Ox对氧化反应催化性能优于α-MnO2。
通过NH3-TPD和CO2-TPD对催化剂的酸碱性质进行分析,结果如图6所示。催化剂表面酸/碱量见表2。133 ℃左右的脱附峰归属于弱酸/碱位上NH3和CO2分子的吸附,而NH3-TPD谱图中较高温度的脱附峰归属于中强Lewis酸性中心。由表2可以看出,与Fe3O4复合后,催化剂Mn8Fe3Ox酸量和碱量均有所增多,酸量由465.2增加至499.5 μmol/g,碱量由11.6增加至12.9 μmol/g。在Mn基氧化物中,碱性中心主要来源于O2−,复合催化剂中碱量的增加进一步说明表面晶格氧O2−相对含量增加[32]。采用Py-FTIR对催化剂酸性质进行分析,结果如图7所示。由分析结果可知,相对Fe3O4和MnO2,复合催化剂Mn8Fe3Ox中波数为1440和1587 cm−1的Lewis酸特征峰强度增大,表明复合催化剂Lewis酸性中心增多,而Mn氧化物中Lewis酸性中心主要是由Mn4+产生,Lewis酸性中心增多是Mn4+含量增加的结果[33]。综上,催化剂酸性质分析结果表明,与Fe3O4复合后,催化剂Mn8Fe3Ox酸性中心和碱性中心数目均有所增加,这是催化剂中Mn4+和O2−相对含量增加的结果,该结果与XPS分析结果一致。
2.3.2 溶剂对HMF氧化反应的影响
Gawade等[34]以MnFe2O4为催化剂进行HMF氧化反应制备DFF,比较了不同溶剂对氧化反应的影响。结果表明,在甲苯等非极性溶剂及水和乙醇等极性质子溶剂中进行的反应效果均不及在1,2-二氧六环和乙腈等极性非质子溶剂。本研究同样比较了不同溶剂对HMF氧化反应的影响。以用量为 0.002 g/mL的 Mn8Fe3Ox为催化剂,将0.6 mmol的 HMF 和 6.5 equiv.TBHP 溶于 5 mL 溶剂中,在80 ℃反应12 h,分析结果见表3。在水和乙醇中,HMF转化率均不及30%,表明此类极性质子溶剂不利于HMF氧化反应的进行。当溶剂为DMF和MeCN时,HMF转化率有所提高,尤其是在MeCN溶剂中获得了26.7%的DFF的收率,这与Gawade研究结果一致,但FDCA收率并不高,呋喃类氧化产物总收率仅为6.8%和57.8%。相对其他几种溶剂,在DMSO溶剂中进行的HMF氧化反应效果最好,转化率高达97.3%,呋喃类氧化产物收率为91.5%。因此,继续以DMSO为溶剂进行反应条件优化。
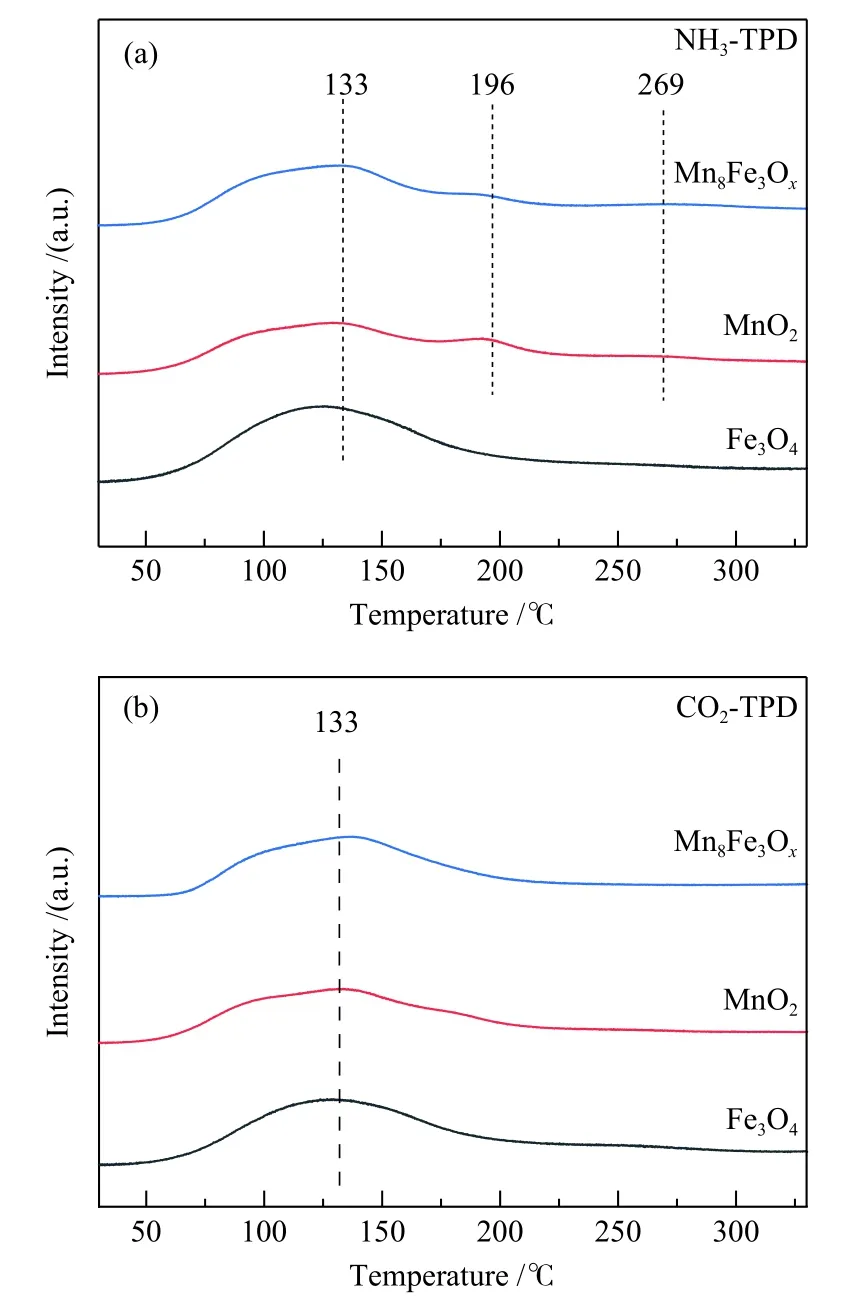
图6 催化剂 NH3-TPD (a)和 CO2-TPD (b)曲线Figure 6 NH3-TPD (a) and CO2-TPD (b) profiles of various catalysts

表2 催化剂的酸量和减量Table 2 The acidity and basicity of various catalysts

图7 催化剂 Py-FTIR 谱图Figure 7 Py-FTIR spectra of various catalysts

表3 不同溶剂对HMF氧化反应的影响Table 3 Influence of different solvents on oxidation of HMF
2.3.3 反应温度对HMF氧化反应的影响
在溶剂DMSO中加入0.002 g/mL的Mn8Fe3Ox催化剂、0.6 mmol的 HMF 和 6.5 equiv.TBHP,在不同温度条件下反应12 h,比较不同温度对HMF氧化反应的影响,结果如图8所示。根据分子动力学理论,反应温度的升高可以提高反应物分子的动力学迁移率,促进反应物分子与催化剂活性位点的接触频率,减小传质阻力,因此,适当升高反应温度有利于反应的进行[17]。但超过90 ℃时,氧化产物FDCA、FFCA和DFF的收率均开始下降,这是由于在高温下,HMF、DFF和FFCA不能保持稳定,生成醚类、乙酰丙酸和不溶性腐殖质等水化和聚合反应[35]。在80 ℃时,中间产物FFCA收率最高,达到43.8%,表明中间产物DFF在此条件下可大部分被继续氧化生成FFCA,但FFCA进一步氧化为FDCA的反应为反应的控速步骤,反应不易进行[15]。继续升高温度,FFCA收率降低,说明升高温度可促进FFCA转化为FDCA,但FDCA在较高温度条件下不能稳定存在,可发生开环反应生成小分子氧化产物,该结果表明升高温度并不能有效增大FDCA收率,只能通过延长反应时间或增大催化剂用量促进中间氧化产物FFCA向FDCA转化。
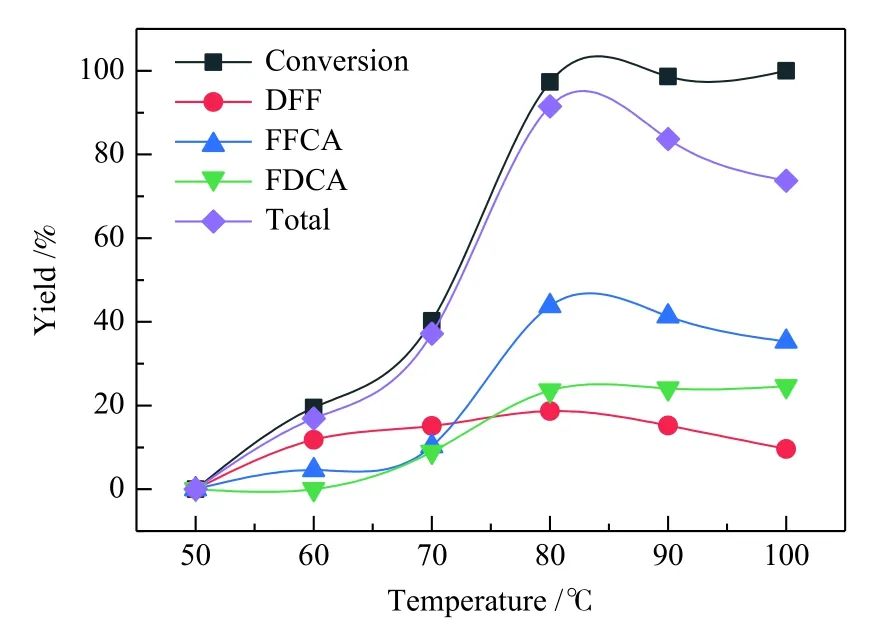
图8 反应温度对 HMF 氧化反应的影响Figure 8 Influence of reaction temperature on HMF oxidation
2.3.4 氧化剂和催化剂用量对HMF氧化反应的影响
在相同反应条件下进行HMF氧化反应,考察氧化剂用量和催化剂用量对反应的影响,结果见表4。HMF完全氧化为FDCA所需氧化剂TBHP理论用量为 3.0 equiv.,但由于 TBHP 在 80 ℃ 易分解,因此,当氧化剂用量为 3.0 equiv.时,HMF 不能被完全氧化,只能得到43.6%的HMF转化率。氧化剂用量增加至6.5 equiv.时,转化率增大至97.3%,FDCA收率为23.6%,继续增加氧化剂用量至9.0 equiv.,HMF虽然完全转化,但FDCA收率和选择性并未提高。因此,6.5 equiv.的氧化剂用量已满足HMF氧化反应需要。
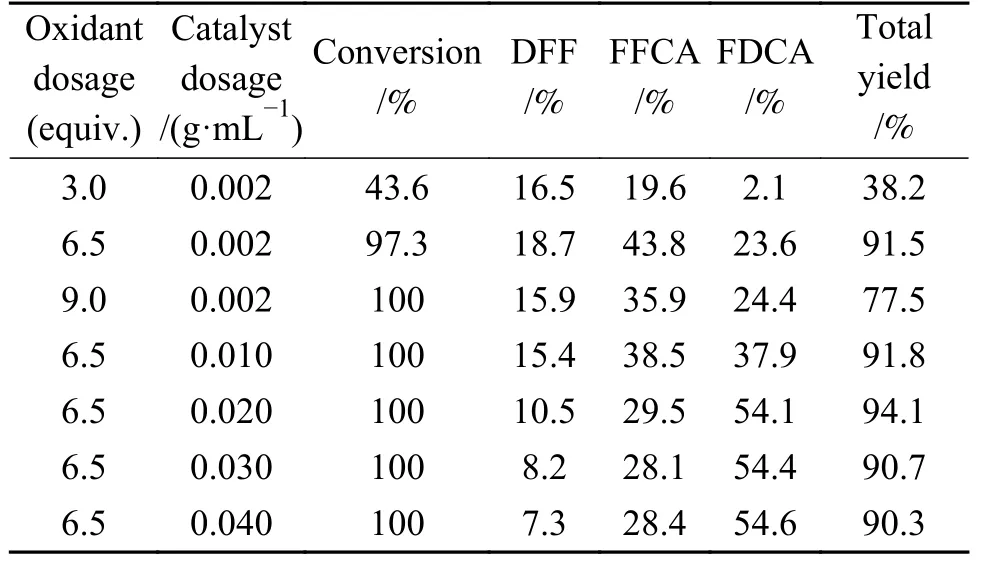
表4 氧化剂和催化剂用量对HMF氧化反应的影响Table 4 Influence of the dosage of oxidant and catalyst on HMF oxidation
由表4还可以看出,催化剂用量对HMF氧化反应的影响较为显著,在 0.002−0.020 g/mL,FDCA收率随催化剂用量的增加显著增大,当催化剂用量为0.002 g/mL时,由于催化活性中心数量较少,FDCA收率仅为23.6%,当催化剂用量增大至0.020 g/mL时,FDCA收率增大至54.1%,继续增加催化用量,FDCA收率并未继续增大,该结果表明0.020 g/mL为HMF氧化反应的最佳催化剂用量。
2.3.5 反应时间对HMF氧化反应的影响
使用最佳用量的氧化剂和催化剂,在80 ℃进行HMF氧化反应,分析不同反应时间对HMF氧化产物分布的影响,结果如图9所示。在80 ℃反应6 h时,HMF即可完全转化,但FDCA收率仅为26.3%,而中间产物FFCA和DFF收率分别为49.5%和16.8%,表明FFCA在6 h内不能全部转化为FDCA。随着反应时间的延长,FDCA收率逐渐增大,中间产物DFF和FFCA收率随之降低,当反应时间延长至24 h时,DFF几乎完全被氧化,FFCA收率也下降至13.9%,而FDCA收率增大至75.5%。继续延长反应时间,FDCA收率并没有随FFCA收率的下降而显著增大,这是由于在此条件下FFCA虽可转化为FDCA,但由于化学性质不稳定,部分FDCA开环分解生成了其他副产物,因此,当反应时间超过24 h后,FDCA收率并未继续增大。
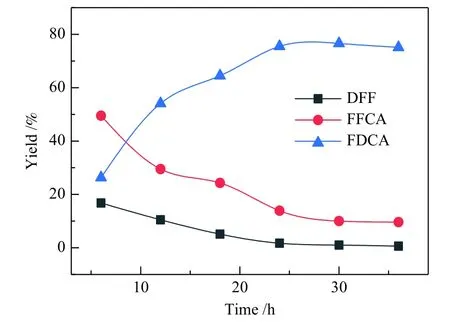
图9 反应时间对 HMF 氧化反应的影响Figure 9 Influence of reaction time on HMF oxidation product yield
2.3.6 正交实验条件优化
在单因素实验基础之上,进一步采用正交实验对HMF氧化反应温度、反应时间、氧化剂和催化剂用量进行条件优化,采用四因素三水平的正交实验,实验方案及结果见表5。正交实验结果表明,各因素对氧化产物FDCA收率的影响顺序为氧化剂用量>反应温度>反应时间>催化剂用量,得出的最有实验条件为C3A1B2D2,即氧化剂用量为9.0 equiv.,反应温度为 70 ℃,反应时间为 24 h,催化剂用量为0.02 g/mL。在此条件下进行HMF氧化反应,氧化产物FDCA收率76.9%。
2.3.7 不同催化剂的催化性能比较
表6将Mn8Fe3Ox催化性能与近年来文献报道的催化剂进行比较,由结果可以看出,Au、Pd、Pt等贵金属基负载催化剂和Fe、Mn、Co等金属的氧化物或复合氧化物对HMF氧化反应表现出较好的催化活性,但反应氧化剂均为高压氧气或高压空气,反应温度较高,且需要大量的碱。而本研究制备的复合金属氧化物催化剂在非碱性条件下于70 ℃即可获得良好的催化效果。
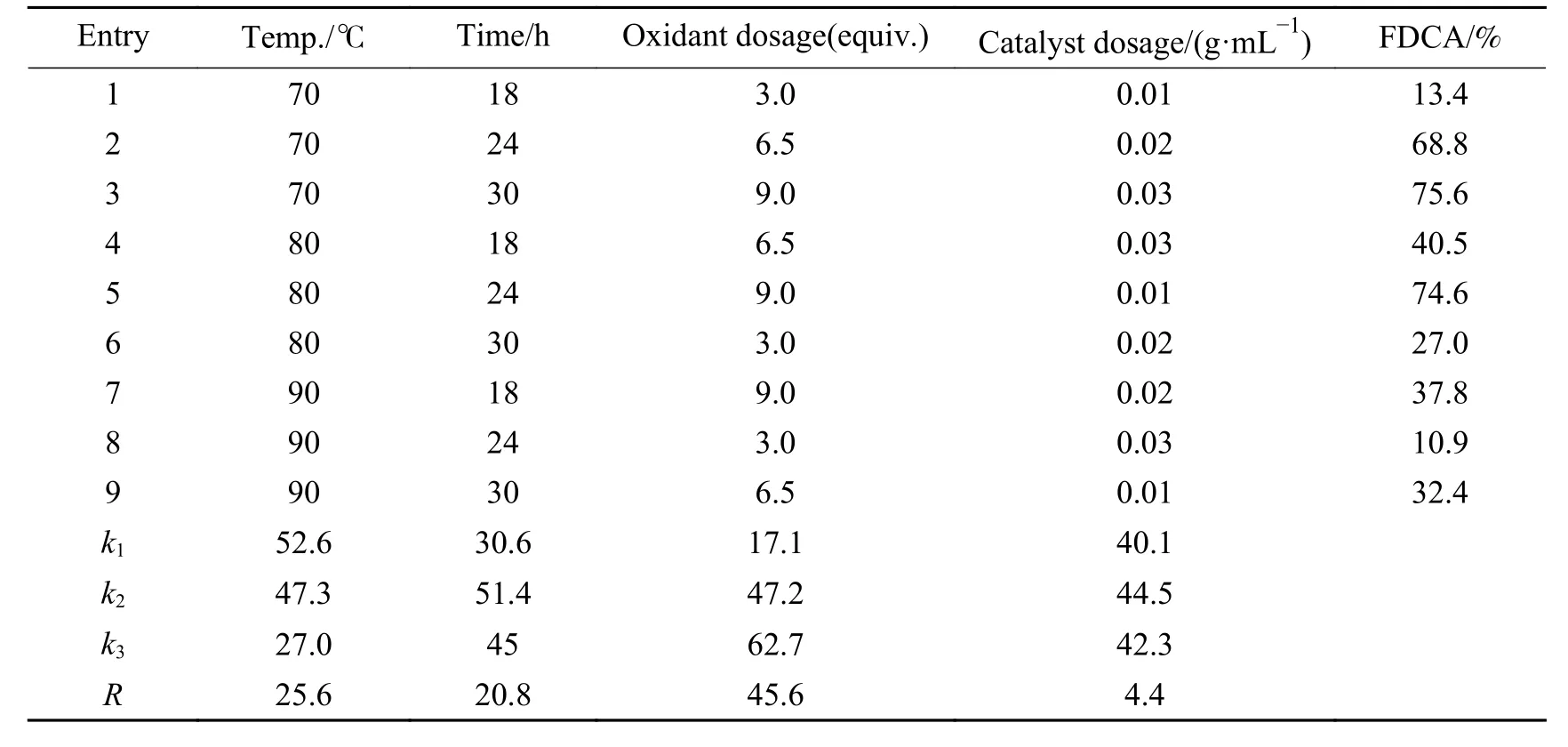
表5 正交结果分析Table 5 Analysis of orthogonal design
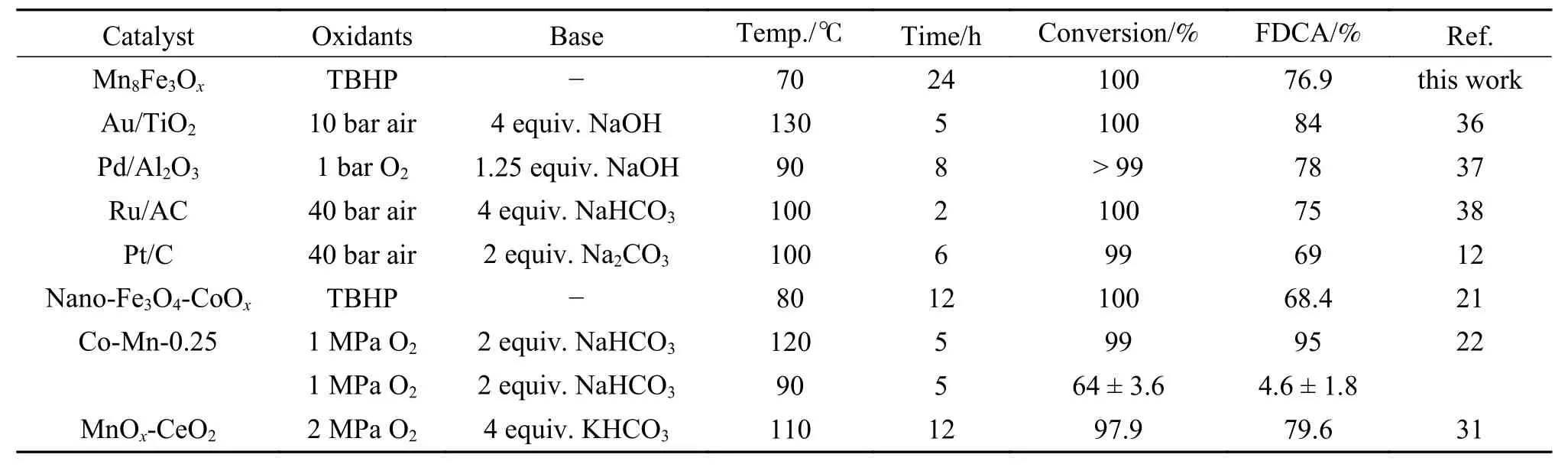
表6 Mn8Fe3Ox催化性能与文献报道其他催化剂比较Table 6 Comparison of the catalytic activity of Mn8Fe3Ox with those of other heterogeneous catalysts reported in the latest literature
非贵金属基催化剂nano-Fe3O4-CoOx在温和条件下对HMF氧化为FDCA的反应也表现一定的催化活性,但反应温度仍高于本研究Mn8Fe3Ox催化HMF氧化反应的最佳温度,且获得的氧化产物FDCA收率与Mn8Fe3Ox也存在一定差距。
2.3.8 催化剂回收与重复使用
反应结束,利用催化剂的磁性将其回收,用水洗涤干燥后重复使用,考察催化剂的重复使用性能,结果如图10所示。
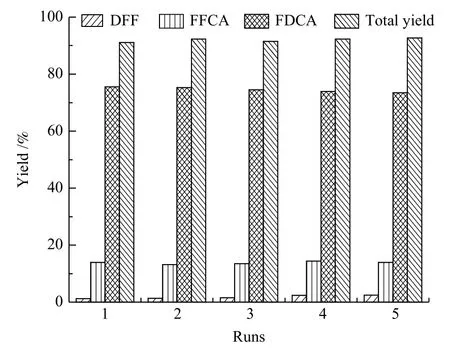
图10 催化剂 Mn8Fe3Ox 重复使用情况Figure 10 Reuse of Mn8Fe3Ox for HMF oxidation
重复使用五次后,HMF仍可完全转化,各氧化产物的收率和呋喃类产物总收率均未显著降低,FDCA收率仍保持在73%以上。该结果表明该HMF氧化反应体系具有良好的可重复性,复合氧化物Mn8Fe3Ox催化剂具有较好的催化稳定性。
3 结 论
将催化活性较好的α-MnO2与Fe3O4复合形成磁性复合氧化物Mn8Fe3Ox,SEM和XRD表征可以看出,复合氧化物Mn8Fe3Ox的形貌和晶型并未发生显著变化。XPS和酸性分析结果表明,与单一氧化物相比,复合氧化物Mn8Fe3Ox中Mn4+和表面晶格氧O2−相对含量增加,使催化活性中心Mn4+·O2−离子对数量增大,这是复合氧化物Mn8Fe3Ox对HMF氧化生成FDCA的反应表现出更好催化活性的主要原因。利用单因素和正交实验法对反应温度、反应时间、溶剂类型、催化剂和氧化剂用量进行优化,结果表明,氧化剂用量是影响HMF氧化反应的最主要因素,在最优化条件下,HMF可完全转化,FDCA收率为76.9%。
猜你喜欢
世界农药(2021年3期)2021-12-09
云南化工(2020年11期)2021-01-14
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
中国食品(2020年9期)2020-05-26
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
分析化学(2017年9期)2017-10-16
浙江农业学报(2017年1期)2017-05-17
烟草科技(2015年8期)2015-12-20
