
化学链燃烧钴基载氧体炉内脱汞机理研究
2021-03-25 01:27倪明国刘敦禹乌晓江
上海理工大学学报 2021年1期
冯 亮, 倪明国, 刘敦禹, 金 晶, 乌晓江
(1.上海理工大学 能源与动力工程学院 上海 200093;2.上海市动力工程多相流动与传热重点实验室,上海 200093;3.上海锅炉厂有限公司,上海 200093)
煤炭燃烧产生的CO2会引起温室效应,为减少CO2排放,提出了多种新型捕集CO2的技术,如:富氧燃烧、燃烧前和燃烧后捕集[1]。其中,整体煤气化联合循环发电(IGCC)、富氧燃烧和化学链燃烧(CLC)技术的目标是采用提高CO2浓度的方法,降低CO2分离成本。CLC 技术可以产生高浓度的CO2气体用以进行捕集、利用或储存,从而减少CO2排放。在CLC 技术中,煤不直接与空气接触,而是在还原反应器中被载氧体(MxOy)氧化。同时,MxOy被还原为MxOy-1,然后被输送到氧化反应器中与氧气反应,并返回到氧化态[2]。通常在CLC 技术中使用的载氧体具有在炉内氧化Hg0的潜力。
如果Hg0可以在炉内高温下被氧化为Hg2+,则Hg2+可能会被粉煤灰吸附而逐渐形成HgP,随后可以通过布袋除尘器和烟气脱硫装置去除HgP。在此过程中,炉中足够长的反应时间可以提高Hg0的脱除效率[3]。目前,Fe2O3已被广泛用作煤的CLC的载氧体,实验结果表明,还原反应器中25%的汞以Hg2+的形式存在,氧化反应器中54%的汞以Hg2+的形式存在。因此,在炉内将Hg0转化为Hg2+是可以实现的,而载氧体Fe2O3是实现Hg0氧化的关键[4]。
目前,已有学者对锅炉中汞的转化规律进行了研究,汞转化的关键因素包括温度、煤种和降温速率等[5]。煤中的汞排放依赖于温度,当温度高于650 ℃时,煤中的汞会随着煤燃烧而完全释放[6]。并且,煤种和炉内停留时间对气相中汞的存在形式也有影响[7]。具体而言,不同的煤中汞和氯的含量对气相汞排放量有很大影响。尤其在高温下,从煤中释放的氯主要以Cl 的形式存在,然后形成HCl 或Cl2。在烟道气体温度降低的过程中,混合烟气中含Cl 物质与Hg0/HgCl 反应可以生成HgCl2[8]。此外,HgCl 到HgCl2的转化速率比Hg0到HgCl 的转化速率快,因此,HgCl 是重要的中间体。在高于600 ℃下,HCl 能够有效地氧化Hg0。同时,在Hg0/O/H/Cl的反应体系中,Cl2对Hg0的氧化速度比HCl 的更快。而Hg0和Cl 之间的反应速度比Hg0/HgCl 和Cl2之间的快[9]。因此,含Cl 组分对Hg0的氧化速度依次为Cl>Cl2>HCl。而且,降温速率对Hg0的氧化也有重要影响,在HCl 质量浓度为300 ppm、降温速率从210 K/s 增加到440 K/s时,Hg0的氧化程度从34%提高到86%[10]。
另一方面,催化剂作用下的Hg0的氧化机理更加复杂。在HCl 气氛下,纳米Fe2O3对Hg0的氧化有显著的促进作用。该催化活性可能归因于迪肯反应,通过形成Cl2促进了Hg0的氧化[11]。此外,在低温(<280 ℃)下,Eley-Rideal 机制是最可能的Hg0氧化途径;在280~580oC 下,Langmuir-Hinshelwood 机理为最有可能的Hg0氧化途径;在高温(>680 ℃)时,Hg0和HCl 之间的均相反应机理以及Langmuir-Hinshelwood 机理均有利于Hg0的氧化[12]。
目前,脱汞的研究缺乏CLC 条件下Hg0在载氧体表面催化氧化的机理,尤其是温度和载氧体的还原对Hg0脱除的影响仍不清楚。同时,飞灰和载氧体的分离也不容忽视[13]。在选择磁性载氧体时,通过比较铁磁矿石(例如Fe3O4,γ-Fe2O3,NiFe2O4和MnFe2O4)和铁磁金属(例如Co,Fe 和Ni)的退磁温度,发现Co 具有最高的退磁温度(1 127 ℃)[14],其他矿物的退磁温度远低于CLC(800~950 ℃)的温度范围[15]。因此,利用钴基载氧体可以解决从煤灰中分离载氧体的困难。在分离过程后,钴基载氧体可以在燃烧过程中重复使用。因此,使用钴基载氧体作为载氧体是具有很好的工业应用价值的。然而,在CLC 条件下,钴基载氧体对Hg0氧化的机理研究很少。有关钴基氧化物催化作用的研究都集中在低温下进行,研究结果表明,Co3+是Co 原子的最高氧化态,在Hg0氧化过程中,Co3+能够夺取Hg0的电子[16]。
本文旨在探讨CLC 中钴基载氧体氧化Hg0的潜力、程度和机理。将Co3O4用作典型的Co 基载氧体,实现在CLC 条件下脱汞的目的。研究了HCl和Hg0在Co3O4表面的催化反应机理,以及温度和载氧体还原对脱汞的影响。本文对炉中Hg0的脱除提供了新的手段,与传统的喷入活性炭粉末脱除汞相比,在整体工艺成本和回收利用的成本方面具有较大优势。由于活性炭脱汞消耗的活性炭量巨大,且使用后很难回收再利用;而使用Co3O4作为氧化汞的载氧体,此过程不需要附加脱除设备,并且由于其具备磁性,可以在炉内使用后利用磁性回收,有利于回收再利用,减少成本。
1 实验装置及方法
1.1 实验装置
图1 是本研究使用的脱汞实验台的系统图。本实验使用小型固定床反应器评价载氧体的脱汞性能。实验系统包括:配气装置(载气瓶)、汞发生装置(汞渗透管、U 型管、水浴锅)、反应装置(管式炉)、汞监测装置(燃煤烟气测汞仪)以及尾气处理装置(活性炭管和碱液洗瓶)。所使用的汞测量仪器为QM201H,该仪器是痕量及超痕量汞检测的专用仪器,其测量范围在0.01~100 μg/L 之间,其检测下限不大于0.003 μg/L,标准曲线的相关系数不小于0.996,重复性及变异系数不大于5%,因此汞测量手段误差较小。在每次实验时,均会使用已标定浓度的标准样先进行检测,每次使用系统中测得的汞浓度与标准样进行对比,时间2 h 左右,当汞蒸气浓度数据波动不大于5%时,再开始进行实验数据的采集。实验参数为:总气体流量为1 L/min,CO 流量 为20 ml/min,200 目Co3O4的添加量为100 mg,加入的石英砂为1 g,NaOH溶液的浓度为0.8 mol/L,汞的质量浓度范围为50±2 μg/m3。
1.2 材料制备及评价
Co 和SiO2购于阿拉丁化学公司。此外,为了获得Co3O4,将50~70 μm 的Co 在700oC 下煅烧1.5 h,然后将其研磨至45~75 μm 的粒径范围[17]。此外,汞测量装置为QM201H 型燃煤烟气汞测量仪,测定单质汞的范围为0~1 mg/m3。在制备Co3O4时,当煅烧温度接近900 ℃时,催化剂严重烧结,并且Co3O4的熔点为900 ℃[18]。因此,将实验温度范围选择为800~890 ℃,在模拟中也使用相同的温度范围。此外,将反应器入口和反应器出口的Hg0浓度表示为Ci和Co。Hg0的氧化效率由下式表示
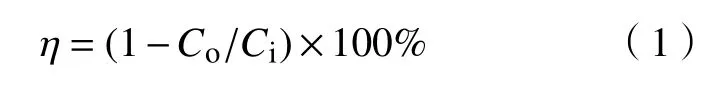
1.3 热力学计算
为了分析Hg0氧化的反应过程,使用FactSage中的Equilib 模块计算化学热力学平衡。在平衡分析中,在给定的压力、温度和系统组成的情况下,当整个系统的吉布斯自由能变ΔG为0 时,意味着系统处于热力学平衡状态。在FactSage 中,Equilib 模块可以帮助判断反应过程。吉布斯自由能G也用作判断在特定温度下可能发生的反应。任何化学反应的吉布斯自由能变都可以用产物的吉布斯自由能变减去反应物的吉布斯自由能变来表示。如果吉布斯自由能变小于零,则意味着化学反应在没有外部能量输入的情况下可以自发产生;如果吉布斯自由能变大于零,则意味着化学反应不能自发进行。
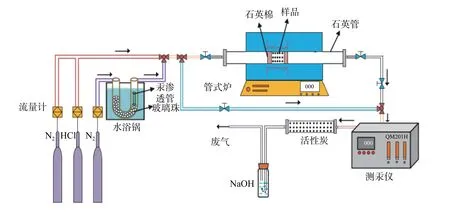
图1 脱汞实验台系统Fig.1 Experimental system of Hg0 removal
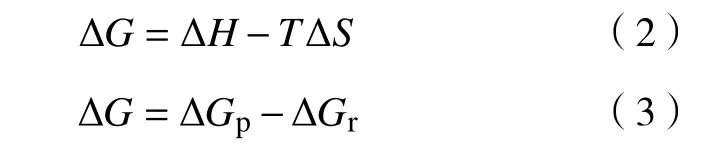
式中:ΔH为焓变;ΔS为熵变;T为温度;ΔGp为产物的吉布斯自由能变;ΔGr为反应物的吉布斯自由能变。
2 脱汞实验
2.1 不同对照组的Hg0 脱除实验
图2 是 SiO2+Hg0、 HCl+Hg0、 Co3O4+Hg0和Co3O4+HCl+Hg0的4 个实验中Hg0的氧化效率图,该实验所使用的HCl 质量浓度为50 ppm。此对照组的结果表明,Hg0的氧化效率遵循以下顺序:Co3O4+HCl+Hg0>HCl+Hg0>Co3O4+Hg0>SiO2+Hg0。
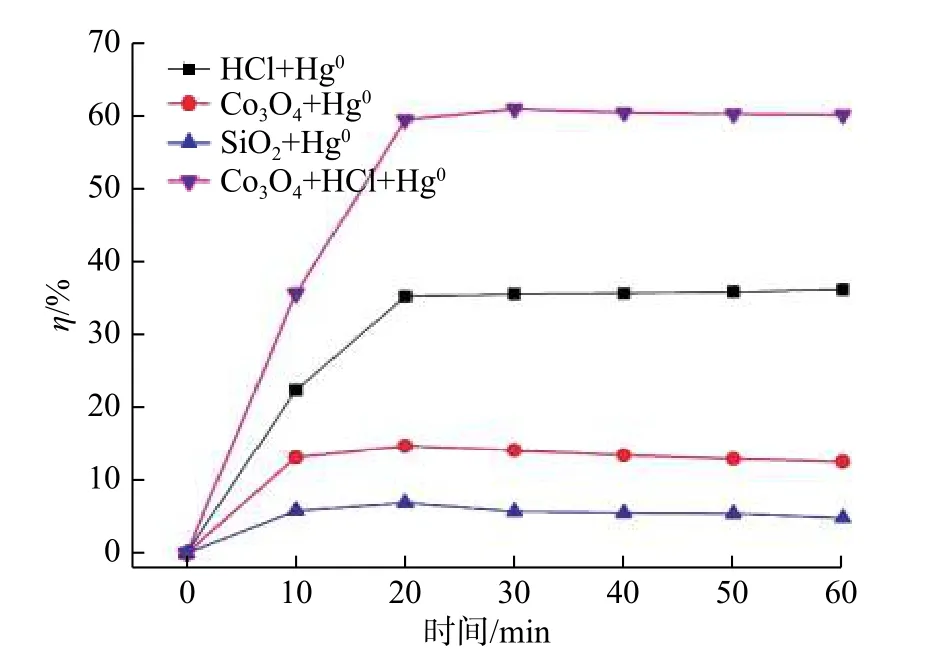
图2 800 ℃下不同条件的Hg0 脱除效率Fig.2 Removal efficiency of Hg0 under different conditions at 800 ℃
实验中将SiO2与Co3O4预混合以使气流更均匀,并防止Co3O4被气流夹带。为验证SiO2对Hg0的氧化效率,进行SiO2+Hg0的实验,实验结果表明,Hg0的氧化效率约为6%,因此,SiO2氧化Hg0的能力很弱。SiO2的脱汞效率可能来自其对Hg0的物理吸附而不是化学吸附。与之对比,在Co3O4+Hg0的实验中,Hg0的氧化效率约为14%,这可能因为Hg0与Co3O4中的晶格氧反应形成HgO[19]。
在HCl+Hg0的反应中,Hg0脱除效率约为36%。在高温下,HCl 氧化Hg0逐渐形成HgCl2。在Co3O4+HCl+Hg0的反应中,Hg0的脱除效率为61%。通过对Co3O4+Hg0和HCl+Hg0实验中脱汞效率的比较,同时加入HCl 和Co3O4可以有效地提高Hg0的脱除效率。这可能由于HCl 能够将Co3O4表面氯化,生成更多的活性中心来氧化Hg0[20]。因此,HCl 有两方面影响:一方面, HCl可以直接氧化Hg0形成HgCl2;另一方面,HCl 可以与金属氧化物反应形成金属氯化物,可以有效地氧化Hg0形成HgCl2[21]。因此,高温下,在Hg0氧化的过程中,均相氧化反应和气固异相氧化反应都很重要。
2.2 高温下的气相和催化氧化反应
Hg0的氧化也和温度相关,本文在CLC 温度范围内,研究了HCl+Hg0和Co3O4+HCl+Hg0的脱汞效率,所使用的HCl 质量浓度为50 ppm。图3是Co3O4+HCl+Hg0和HCl+Hg0两组试验在800~890 ℃的温度范围内的脱汞效率图。随着温度的升高,Co3O4+HCl+Hg0的脱汞效率从61%增加到78%,HCl+Hg0的脱汞效率从36%逐渐增加到66%。且随温度的升高,HCl+Hg0的脱汞效率与Co3O4+HCl+Hg0的脱汞效率越来越接近,证明HCl和Hg0的均相氧化反应对脱汞效率的影响随温度的增加而增强。与此同时,Co3O4对HCl 和Hg0反应的影响降低。尽管如此,在相同温度下,Co3O4+HCl+Hg0实验中Hg0的脱除效率仍高于HCl+Hg0实验中Hg0的脱除效率,这说明Co3O4的催化氧化作用不可忽略。
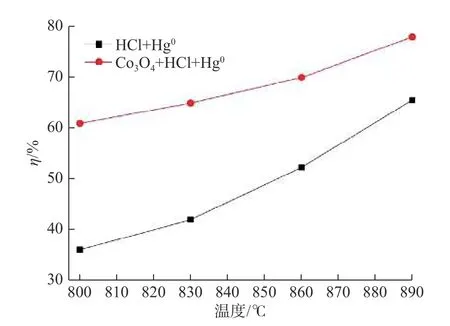
图3 气相反应(HCl+Hg0)和催化氧化(Co3O4+HCl+Hg0)的脱汞效率Fig.3 Removal efficiency of Hg0 for gas-phase reaction(HCl+Hg0)and catalytic-oxidation reaction (Co3O4+HCl+Hg0)
2.3 还原性气氛下Hg0 的氧化
图4 是不同反应体系的Co3O4+Hg0,Co3O4+HCl+Hg0,20 minCO+Co3O4和40 minCO+Co3O4的X 射线衍射(X-ray Diffraction,XRD)图谱。其中,20 minCO+Co3O4是指Co3O4样品被CO 还原20 min;类似地,40 minCO+Co3O4是指Co3O4样品被CO 还原40 min。从Co3O4+Hg0的XRD 图可以看出,反应过后,产物组成仍为Co3O4,相应实验中Hg0的氧化效率为14%(如图2 所示),这说明与Hg0反应的Co3O4的量很少。Co3O4+HCl+Hg0的XRD 图显示,产物中有CoO,并且Co3O4的特征峰消失,其结果可解释为,在反应过程中HCl 消耗了大量Co3O4。由于HCl 和Co3O4之间的反应形成了含Cl 的物质,Hg0+Co3O4+HCl 中Hg0的氧化效率为61%(如图2)。由于系统中主要产物是CoO,因此Co3O4主要转化为CoO。40 min CO+Co3O4的XRD 图显示产物中的成分为Co,Co3O4的特征峰消失了。这表明Co3O4被完全还原为Co。 20 minCO+Co3O4的XRD 图 显 示Co 和CoO 共存。因此,还原过程路径为:Co3O4首先被还原为CoO;然后被还原为Co。
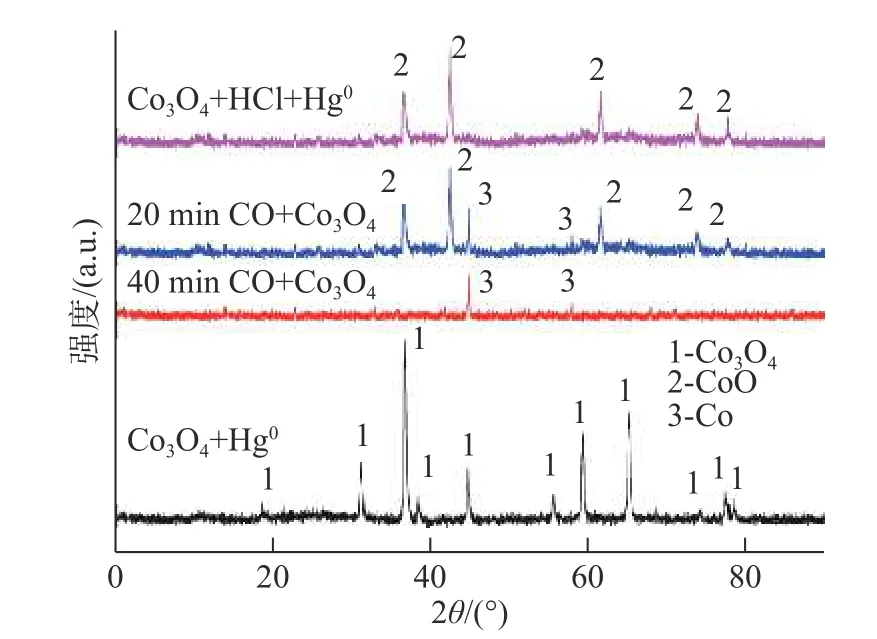
图4 Co3O4+HCl+Hg0 和Co3O4+Hg0 在不同实验条件下的载氧体XRD 图谱Fig.4 XRD patterns of the contrasted oxygen carriers under different conditions with Co3O4+HCl+Hg0 and Co3O4+Hg0
图5 是先使用20 ml/minCO 处理Co3O4样品,将其在不同还原时间的产物,放置在50 ppmHCl+Hg0气氛下进行脱汞实验。对于20 minCO+Co3O4来说,Co3O4被CO 还原20 min 后,其还原产物在50 ppmHCl+Hg0系统的脱除效率为39%,20 minCO+Co3O4的XRD 图显示样品组成为Co3O4和CoO,即当部分Co3O4被还原为CoO 后,50 ppmHCl+Hg0系统的脱汞效率下降。与图2 对比可以看出,50 ppmHCl+Co3O4+Hg0的脱汞效率为61%,这意味着Hg0的脱除效率随着Co3O4的还原程度的增加而降低。因此,Co3O4对Hg0的催化氧化能力优于CoO。另外,40 minCO+Co3O4的XRD图显示样品中只含有Co,此时,该还原样品在50 ppmHCl 气氛中,Hg0脱除效率为35%,低于被还原20 min 样品在50 ppmHCl+Hg0系统的脱汞效率39%。因此,Co3O4及其还原产物对Hg0脱除能力的顺序为Co3O4>CoO>Co。
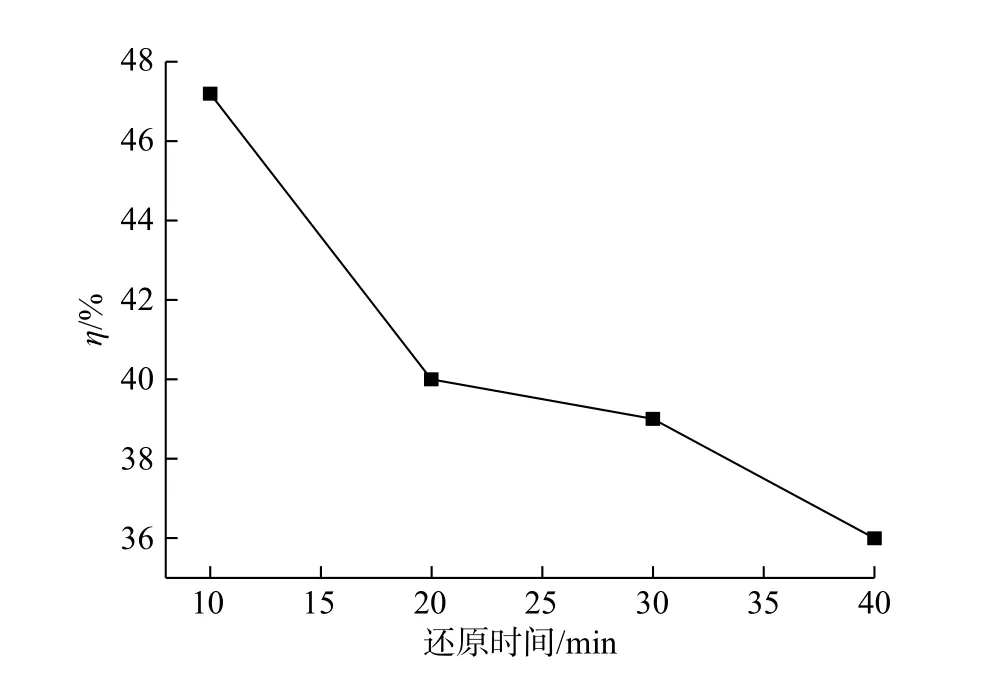
图5 50 ppm HCl 气氛下,Co3O4 不同还原时间产物的脱汞效率Fig.5 Hg0 oxidation efficiency of Co3O4 with different CO reduction times under 50 ppm HCl atmosphere
3 Hg0 氧化机理
3.1 Hg0 氧化机理的实验结果
图6 为不同方法预处理样品的Hg0脱除效率,该实验中所使用的HCl 质量浓度为50 ppm。对于Hg0+HCl/Co3O4,其实验过程为:首先将HCl 引入石英管中,与Co3O4反应2 h;然后用N2冲洗HCl/Co3O4样品0.5 h;最后将样品用于Hg0脱除实验[22]。
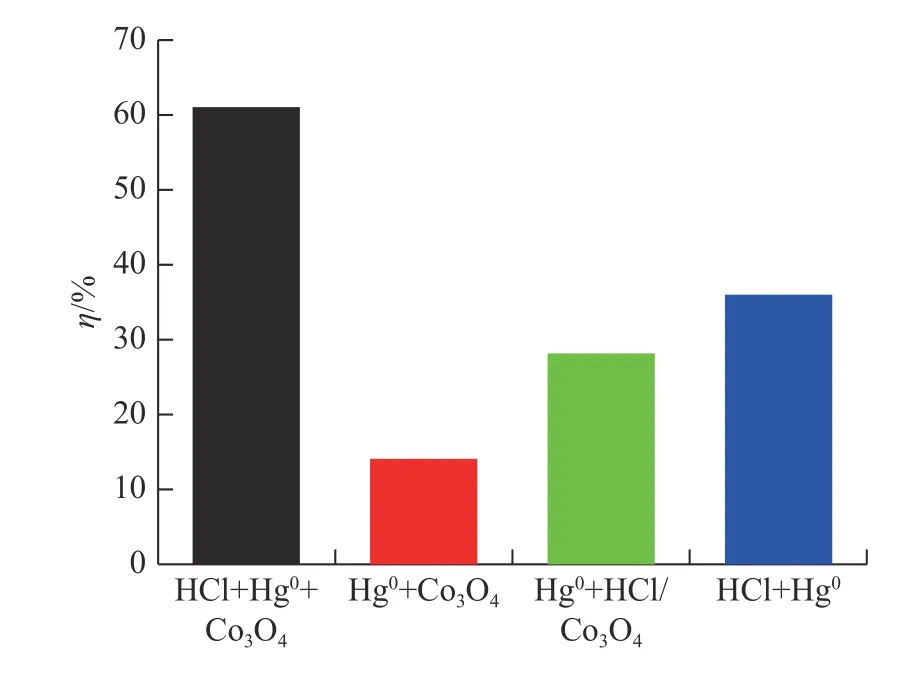
图6 不同对照组实验的脱汞效率Fig.6 Hg0 removal efficiencies for experiments with different control groups
Hg0+Co3O4中Hg0脱除效率仅为14%,而HCl+Hg0+Co3O4中Hg0脱除效率显著地提高到61%。产生较大差异的原因为Hg0在Co3O4表面的吸附并不是Hg0脱除的主要反应途径。同样,Hg0+HCl/Co3O4系统的Hg0脱除效率为28%,这意味着HCl在Co3O4表面的预吸附并未实现61%的Hg0脱除效率,HCl 预吸附也不是主要的反应途径。同时,在Hg0与HCl 的均相反应路径中,Hg0的脱除效率为36%,因此,均相反应对Hg0脱除的贡献很大。排除上述吸附方式后,脱汞机理可能为HCl和Hg0都预吸附在Co3O4表面,然后发生反应实现了Hg0的氧化脱除。同时,Hg0与HCl 的均相反应也有利于Hg0的脱除,气固异相反应的过程可以采用R1-R4 表示。
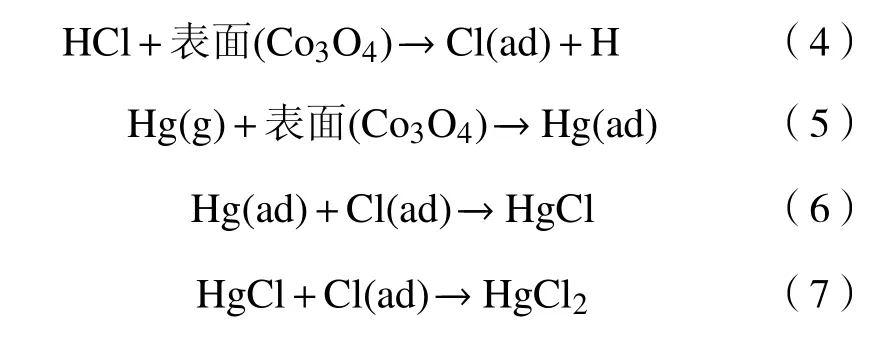
3.2 基于热力学计算的Hg0 氧化机理
3.2.1 鉴定Hg0氧化的重要反应物
对HCl+Co3O4和HCl+Co3O4+Hg0体系均进行热力学计算,通过比较两组的平衡产物,可以确定实现Hg0氧化的重要反应物。对于HCl+Co3O4+Hg0体系,输入参数包含4.150×10-10mol Co3O4,1.996×10-10mol Hg0和2.230×10-6mol HCl。其中,Hg0和HCl 的含量实验条件确定,而Co3O4的含量参考刘亭等[12]的研究。对于HCl+Co3O4体系的模拟,输入参数包含4.150×10-10mol Co3O4和2.230×10-6mol HCl。
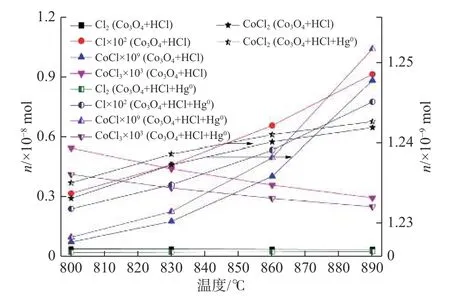
图7 不同温度下Co3O4+HCl 和Co3O4+HCl+Hg0 体系的产物变化Fig.7 Changes of products for two reaction systems of Co3O4+HCl and Co3O4+HCl+Hg0 at different temperatures
图7 为HCl+Hg0和Co3O4+HCl+Hg0反 应 体系,温度从800 ℃升高到890 ℃时不同产物含量的变化。两反应体系的主要产物包括:Cl,Cl2,CoCl,CoCl2和CoCl3。热力学计算表明了CoCl 和CoCl3的存在,而图4 中样品的XRD 图显示样品中不存在金属氯化物,这可能是实际含量太低导致。
为确定对Hg0的氧化有重要影响的物质,比较了两个体系中主要产物的含量。在相同温度下,Co3O4+HCl+Hg0中 的Cl2,Cl 和CoCl3的 量 少 于Co3O4+HCl 中相应成分的含量,而Co3O4+HCl+Hg0中的CoCl 和CoCl2的含量超过Co3O4+HCl 中的相应物质含量。两个体系之间的区别是将Hg0添加到了Co3O4+HCl+Hg0中。该结果表明,添加的Hg0消耗了Cl2,Cl 和CoCl3,促进了Hg0的氧化。而且,Co3O4+HCl 和Co3O4+HCl+Hg0中Cl2/Cl 的含量均随温度的升高而增加,这可以解释图3 中Hg0脱除效率随着温度增加而上升的趋势。
为量化Cl2,Cl 和CoCl3对Hg0氧化的贡献,对Co3O4+HCl 和Co3O4+HCl+Hg0体系进行了含Cl组分的质量平衡计算。根据两体系中Cl 的质量守恒方程,将Hg0添加到Co3O4+HCl 中后,含Cl 组分(2×Cl2,Cl 和3×CoCl3)的减少量应等于含Cl 组分(2×HgCl2,HgCl,CoCl 和2×CoCl2)的增加量。
图8 显示了在Co3O4+HCl 体系中添加Hg0含Cl 组分的减少量和含Cl 组分的增加量。加入Hg0后,在800,830,860,890 ℃下,含Cl 组分减少量与含Cl 组分增加量的百分比例分别为99.3%,98.8%,98.1%和96.8%。因此,可以大致实现含Cl 组分的质量守恒。其中,Cl2,Cl 和CoCl3是减少的主要含Cl 物质;HgCl2,HgCl,CoCl 和CoCl2是增加的主要含Cl 物质。含Cl 组分对Hg0氧化的贡献顺序为:Cl2>Cl> CoCl3。而且,Cl2是Hg0氧化最重要的反应物,HgCl2是Hg0氧化最重要的产物。
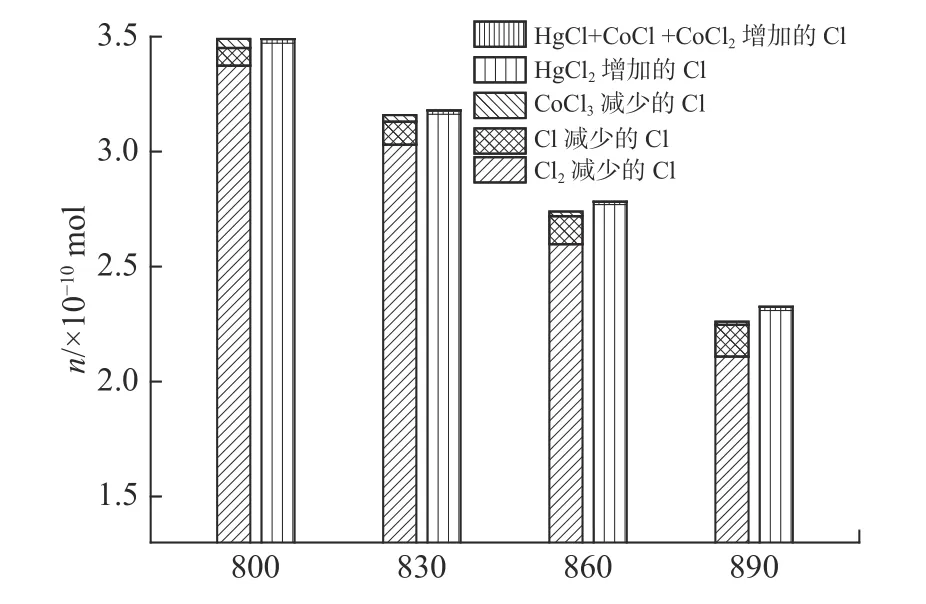
图8 Co3O4+HCl 系统加入Hg0 后减少和增加的含Cl 物质量Fig.8 The reduced Cl and increased Cl-containingcomponents after the addition of Hg0 in the Co3O4+HCl system
3.2.2 含氯物质的产生
基于热力学计算,与Hg0氧化有关的含Cl 组分包括Cl,Cl2,CoCl,CoCl2和CoCl3。为了找到Hg0氧化的确切路径,对可能的基元反应吉布斯自由能进行计算,如表1 所示。对Cl 成分的产生有如下路径。在高温下,HCl 通过反应R5 分解为H 和Cl。由于足量的HCl,此过程会产生大量的H 和Cl。然后两个Cl 通过反应R7 产生Cl2。此外,Co3O4可以与H2通过R9 生成Co 和H2O,然后Co 可以与Cl 通过R12 生成CoCl,CoCl 被氧化为CoCl3。
3.2.3 Hg0被含氯组分氧化
通过计算Cl2/Cl/CoCl/CoCl2/CoCl3和Hg0之间化学反应的吉布斯自由能变,可以确定可能发生的化学反应。含氯组分Cl 和Cl2可以将Hg0逐渐氧化为HgCl2[23]。在此过程中,HCl 与Hg0之间的反应可形成中间产物HgCl,接着与HCl 进一步反应生成HgCl2。
表1 显示了系统中20 个反应的吉布斯自由能变,其中包括含Cl 组分氧化Hg0反应方程式:Cl2+Hg0,Cl+HgCl,Cl+Hg0,Cl2+2HgCl,CoCl3+HgCl和2Cl+Hg0。含氯组分具有极强的氧化Hg0的能力,Cl2被认为是Hg0氧化过程中的重要元素。Laudal 等[24]发现体积浓度10 ppm 的Cl2可以达到84.8%的Hg0氧化效率,而当Cl2的体积浓度高于50 ppm 时,可以达到95%的Hg0氧化效果。考虑到电子结构,Cl2的氧化能力比HCl 强得多。
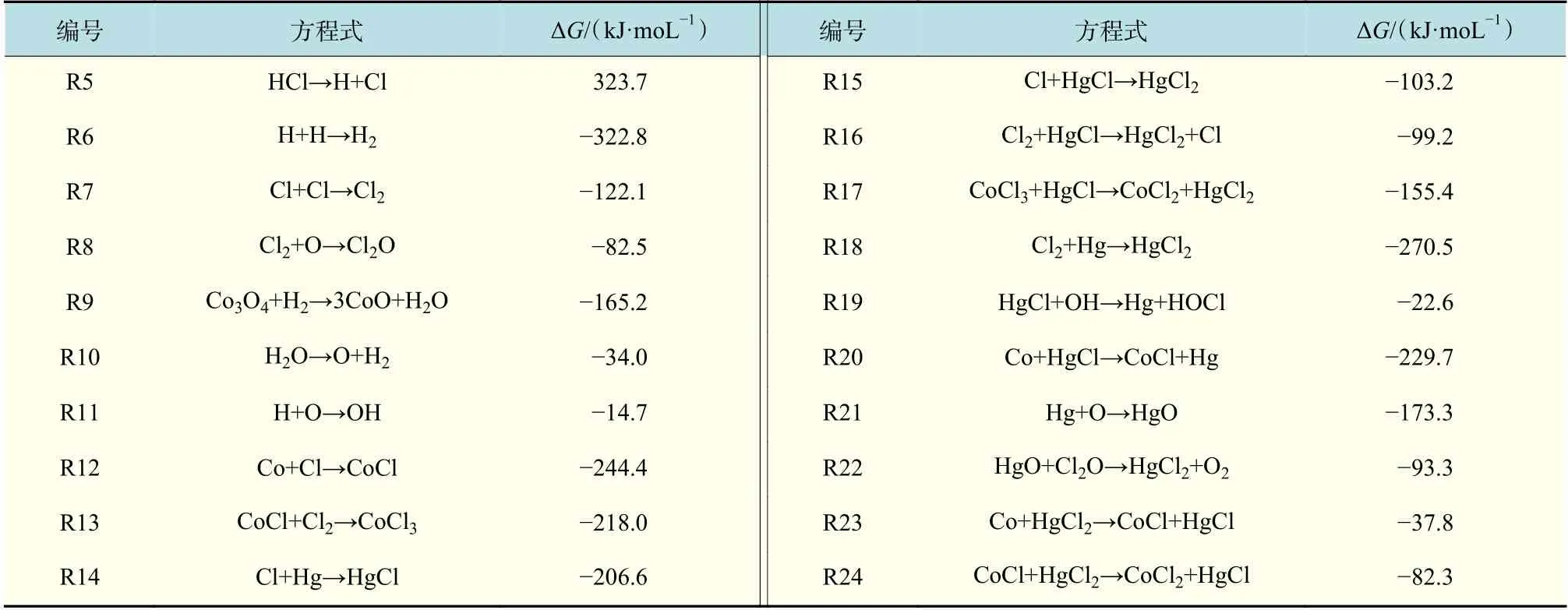
表1 20 个反应方程式在800 ℃下的吉布斯自由能变值Tab.1 Changes of Gibbs free energy for different reactions at 800 ℃
3.2.4 金属氧化物对汞的氧化
Hg0与Co3O4(110)表面之间的反应是化学吸附,吸附能为-74.037 kJ/mol[14]。此外,Co3+是直接的相互作用位点,在此过程中会形成稳定的Hg0吸附系统。反应过程可能如下:Hg0吸附在Co3O4的表面,形成吸附态的Hg0(ad)。然后,Hg0(ad)与氧反应生成HgO。结合上述对于Hg0的氧化途径,表1 显示了800 ℃下,20 个化学反应的吉布斯自由能变。
3.3 机理总结
表1 为在800 ℃下的吉布斯自由能变值,其中19 个反应的吉布斯自由能变均为负,证明这些反应可以自发进行。
上述计算结果表明,Hg0的氧化存在3 种反应路径:在第一种反应路径中,Hg0与Cl2反应直接生成HgCl2;在第二种反应路径中,Hg0与Cl 反应形成HgCl,然后被Cl/Cl2氧化形成HgCl2;在第三种反应路径中,Hg0与O 反应生成HgO,HgO与Cl2O 反应生成HgCl2。图9 为Hg0氧化完整机理图。
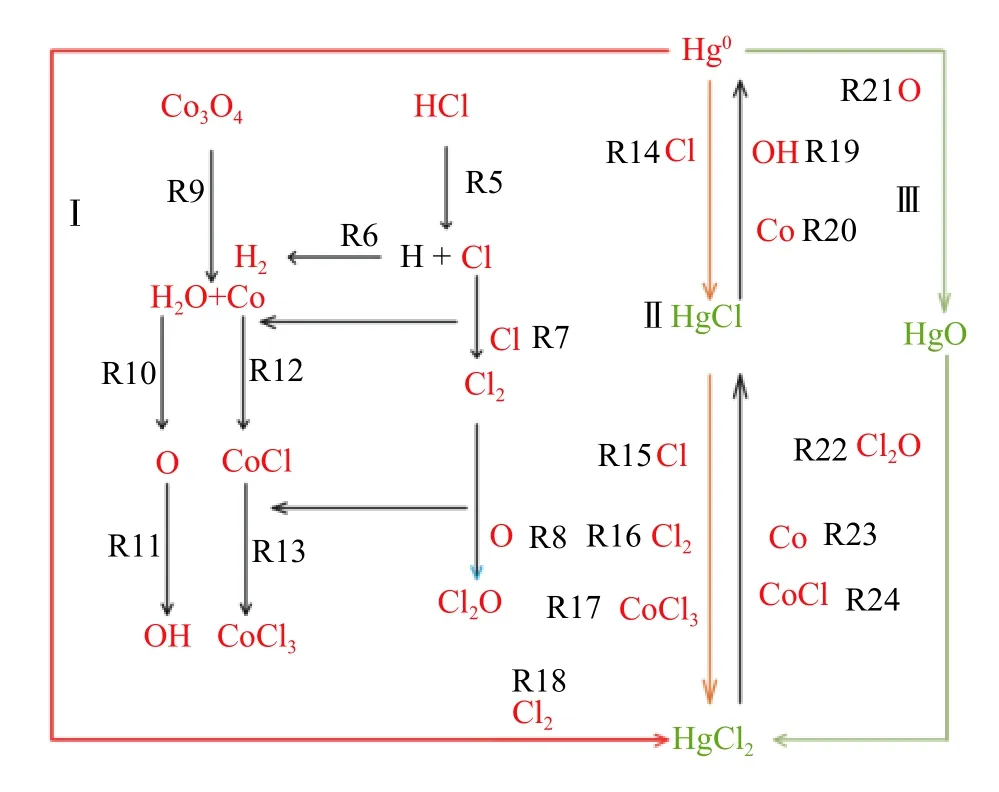
图9 Hg0 的氧化过程相关的机理Fig.9 The relevant mechanism for Hg0 oxidation process
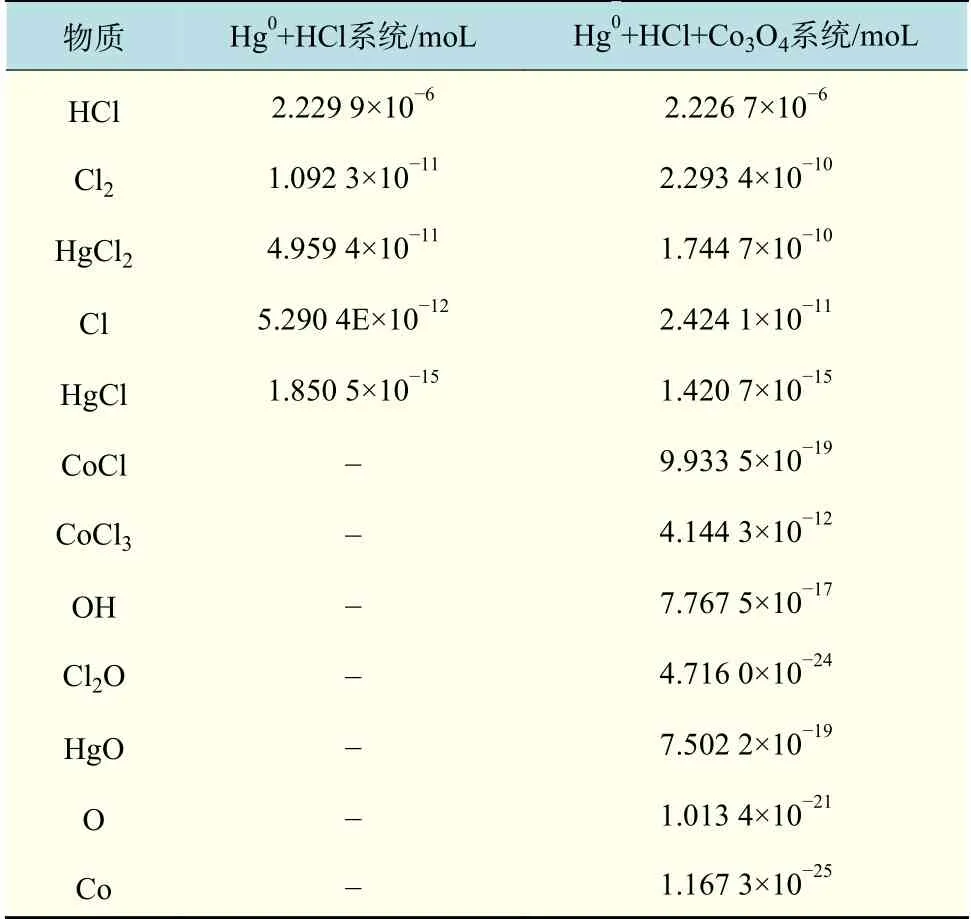
表2 Hg0+HCl+Co3O4 和Hg0+HCl 系统物质的量对比Tab.2 Comparison of reaction produts materials in Hg0+HCl+Co3O4 and Hg0+HCl+Co3O4 system
在氧化Hg0的3 种路径中,对于第一种反应路径,Hg0+HCl+Co3O4和Hg0+HCl 相比,Cl2的量增多,促进了Hg0向HgCl2的转化。Hg0与Cl2反应生成HgCl2(R18)的吉布斯自由能变为-2.7×102kJ/moL,吉布斯自由能变为负值,此反应可以自发进行。在第二种反应路径中,Hg0+HCl+Co3O4和Hg0+HCl 相比,Cl 和Cl2的量增多,促进了Hg0向HgCl2的转化。对于该路径中的还原反应,由于CoCl 和OH 物质的量小于氧化反应过程中Cl 和Cl2的量,所以此过程促进Hg0向HgCl2的转化。Hg0与Cl 反应生成HgCl(R14)的吉布斯自由能 变 为-2.066×102kJ/moL,HgCl 与Cl 反 应 生 成HgCl2(R15)的吉布斯自由能变为-1.036×102kJ/moL,两个反应的吉布斯自由能变为负值,则这两个反应均可以自发进行。在第三种反应路径中,Hg0与O 反应生成HgO,HgO 与Cl2O 反应生成HgCl2。 Hg0+HCl+Co3O4和Hg0+HCl 相 比, O 和Cl2O 的量也会促进Hg0向HgCl2的转化。Hg0与O 反应生成HgO(R21)的吉布斯自由能变为-1.733×102kJ/moL, HgO 与 Cl2O 反 应 生 成HgCl2(R22)的吉布斯自由能变为-9.33×101kJ/moL,两个反应的吉布斯自由能变为负值,均可以自发进行。
4 结 论
使用Co3O4作为氧载体,在CLC 条件下进行脱汞研究,揭示了HCl 和Hg0在Co3O4表面的催化氧化机理。此外,还详细地探讨了温度和氧载体还原对脱汞的影响。得到如下结论:
a.在高温下,HCl 和Hg0在Co3O4表面的催化氧化时,Langmuir-Hinshelwood 机理和均相反应机理对于Hg0的脱除都很重要。
b.Co3O4被CO 还原的途径为:Co3O4→CoO→Co。随着Co3O4还原度的增加,其还原产物对Hg0的脱除效率逐渐降低。
c.在Co3O4与Hg0和HCl 同时反应时,存在3 个反应路径:在第一种反应路径中,Hg0与Cl2反应直接生成HgCl2;在第二种反应路径中,Hg0与Cl 反应形成HgCl,然后被Cl/Cl2氧化形成HgCl2;在第三种反应路径中,Hg0与O 反应生成HgO,HgO 与Cl2O 反应生成HgCl2。
猜你喜欢
煤气与热力(2021年12期)2022-01-19
建材发展导向(2021年14期)2021-08-23
粉末冶金技术(2021年3期)2021-07-28
环境卫生工程(2021年3期)2021-07-21
当代陕西(2020年23期)2021-01-07
英语文摘(2020年7期)2020-09-21
当代化工(2019年3期)2019-12-12
今日财富(2017年32期)2017-10-19
火炸药学报(2014年1期)2014-03-20
火炸药学报(2014年1期)2014-03-20
