
泡桐维管形成层及木质部区的转录组分析
2021-03-22 09:21邱宗波王稳利张雪如
植物研究 2021年3期
滕 云 邱宗波 王稳利 张雪如
(1. 信阳农林学院园艺学院,信阳 464000;2. 河南师范大学生命科学学院,新乡 453007)
林木的次生生长是多年生木本植物维管形成层周期性分裂,向外分化出韧皮部,向内分化出木质部的过程[1]。维管形成层细胞的增殖和分化使树木不断增粗,但在不同类型以及不同地区的树木中有着不同的活动样式。对于温带的木本植物来说,冬季形成层由于低温进入休眠,到次年春天气温回升时开始萌动,夏季形成层由于高温进入旺盛的活跃期[2]。维管形成层随季节由休眠期到萌动期再到活跃期,再由活跃期到休眠期的循环变化在树木的生长和发育过程中起着关键的作用[2~3]。
随着高通量测序技术的发展,转录组(RNASeq)测序已成为了解植物在某一发育阶段或功能状态下基因功能及基因结构的重要手段[3~4]。曹玉婷等[5]利用高通量RNA-Seq 测序技术对4 个不同发育阶段的柳杉维管形成层进行转录组测序,获得了大量与木材形成相关的重要功能基因。杨光等[6]对柚木边材进行转录组测序,初步明确了部分编码蛋白的功能、分类及代谢通路,为柚木分子育种工作的开展提供数据和参考。然而泡桐维管形成层及木质部区组织转录组测序的相关研究尚未见报道。
泡桐(Paulownia spp.)为玄参科(Scrophulariaceae)泡桐属(Paulownia)树种,具有生长快和用途广等优点,是我国重要的速生优质用材树种之一[7]。泡桐中已发现的基因大多是与抗病害相关的基因,与木材材质和产量形成相关基因的挖掘还相对比较薄弱[8]。因此,本研究利用HiSeqTM2500 测序平台对泡桐休眠期、萌动期和活跃期形成层及木质部区组织的混合样品进行了转录组测序和组装,并利用生物信息学的方法,对相关基因进行功能注释和分析,从而为泡桐功能基因的发掘利用,揭示木材形成的分子机制提供理论依据。
1 材料与方法
1.1 植物材料
实验材料取自河南农业大学树木园内生长良好树龄大约为20 年的泡桐。取材时间分别为2018年1月27日(休眠期)、2018年3月18日(萌动期)、2018 年6 月4 日(活跃期),每个时间点选取3株生长一致的泡桐作为试材,分别在1.3 m 树高处用锋利的凿子去除树皮,然后用小刀刮取形成层,迅速装入无RNA 酶的Eppendorf离心管中,用液氮速冻后储存在-80℃冰箱中。
1.2 RNA的提取、文库的制备及测序
采用Plant RNA Reagent 试剂(Invitrogen),按照说明书分别提取泡桐休眠期、萌动期和活跃期维管形成层及木质部区的总RNA。总RNA 样品的质量检测是利用Agilent 2100 生物分析仪(Agilent Technologies,USA)进行的。休眠期、萌动期和活跃期泡桐形成层及木质部区组织的总RNA分别检测合格后,将3 个时期的RNA 等量混合用于测序文库构建。cDNA 文库的制备参照贾新平等[9]方法。建好的文库利用北京诺禾致源生物公司Illumina HiSeqTM2500测序平台进行转录组测序。
1.3 数据组装及功能注释
将测序得到的原始序列(raw reads)去除接头、poly-N 序列、小于20 nt 的序列以及低质量测序片段后,得到高质量干净数据(clean reads)。利用Trinity 软件(参数设置K-mer=25)[10]对clean reads进行De novo 拼接组装,得到长度不一的转录本(Transcripts),并从中选取最长的转录本作为泡桐的单基因簇(Unigene)。通过Blastx 软件[11]将泡桐的Unigenes 序列分别与Nr(Non-redundant protein database)、GO(Gene ontology)、Nt(Non-redundant nucleotide sequences)、SwissProt(SwissProt protein database)、COG(Clusters of orthologous group)、Pfam(Protein families database)和KEGG(Kyoto encyclopedia of genes and genomes)数据库进行比对,从而得到泡桐Unigenes 的功能注释信息。
1.4 泡桐SSR位点分析
使用MISA 软件(http://pgrc.ipk-gatersleben.de/misa/misa.html)从泡桐Unigenes 中搜索简单序列重复(SSR)位点,搜索标准参照许蔷薇等[12]方法。
2 结果与分析
2.1 泡桐RNA-Seq测序及转录组组装
为了获得尽可能丰富的泡桐转录组信息,选取泡桐维管形成层的休眠期、萌动期和活跃期材料分别提取总RNA,然后将这3 个组织的RNA 等量混合后进行转录组测序。经IllluminaTMHiSeq 2500 高通量平台测序,总共获得111 645 144 条原始序列(raw reads),将原始数据进行过滤后,得到109 021 918 条clean reads(约16.35 Gb)。原始数据已上传到NCBI 中的GSE 数据库(登录号为GSE109323)。另外Q20为97.77%,Q30为94.30%,表明测序质量较好,可为后续的序列组装提供高质量的原始数据。
测序得到的clean reads 利用Trinity 软件进行De novo组装,得到145 881个转录本(Transcripts),平均长度为888 nt(见表1)。其中,长度在1 000 nt以下的Transcripts 有104 177 条,占71.41%;长度在1 000~2 000 nt 的有24 155 条,占16.56%;而长度在2000 nt 以上的仅有17 549 条,占12.03%。进一步对转录本聚类去冗长得到104 432 个Unigenes,平均长度为662 nt(见表1)。其中,长度在1 000 nt 以下的Unigenes 有86 607 条,占82.93%;长度在1 000~2 000 nt 的有10 848 条,占10.39%;而长度在2 000 nt 以上的仅有6 977 条,占6.68%。Transcripts 和Unigenes 的N50 分 别 达 到1 655 和1 105 nt,表明组装片段的完整性是比较高的。

表1 泡桐转录组组装结果Table 1 Summary of transcriptome assembly for P.tomentosa
2.2 Unigene基因功能注释及分类
为了获得Unigene 的功能注释信息,我们将组装得到的104 432 个Unigenes 分别在Nr、Nt、SwissProt、GO、COG、KEGG和Pfam 数据库中利用blastx软件进行搜索比对。从表2 可以看出,Nr 数据库注释的Unigenes最多(40 789条),占总Unigenes的39.05%;仅有15 539 条Unigenes 在COG 数据库得到注释,占总Unigenes 的14.87%。Nt、SwissProt、GO、KEGG 和Pfam 分 别 注 释 到31 675、30 499、29 168、16 316、28 828条Unigenes。另外,有48 214条(46.17%)泡桐Unigene 序列可以获得至少1 个数据库的功能注释,而且在7个数据库中都能得到注释的Unigenes 共有7 386 个,占总Unigenes 数的7.07%。
2.3 Unigene的GO功能分析
泡桐Unigene 通过与GO 数据库比对分析,可将注释的29 168个Unigene分为分子功能(molecu-lar function)、生物过程(biological process)和细胞组分(cellular component)3 大类55 个功能组(见图1)。其中,归于生物过程的Unigene 有69 943 个,38 276个Unigene归于细胞组分,33 316个Unigene归于分子功能,这一Unigene 功能注释分类结果显示了泡桐维管形成层发育过程中基因表达的总体情况。在生物过程这一功能分类中,细胞过程(cellular process)(73 324 个)、代谢过程(metabolic process)(162 580 个)和单一有机体过程(single-organism process)(28 373个)功能组注释到的Unigene 数量最多,分别占69 943 条生物学过程Unigene的22.74%、22.02%和16.68%,而生物相(biological phase)注释到的Unigene 最少,仅占0.03%。在细胞组分这一功能分类中,细胞(cell)(19 186 个)和细胞组分(cell part)(30 231 个)功能组注释到的Unigene 数量最多,分别占38 276 个细胞组分Unigene 的20.05%和20.04%,突触部分(synapse part)、突触(synapse)、胞外基质成分(extracellular matrix component)、共质体(symplast)4 个功能组注释到的Unigene 均占38 276 个细胞组分Unigene的0.01%。在分子功能这一功能分类中,结合(binding)(49 522 个)和催化活性(catalytic activity)(12 555个)功能组注释到的Unigene 数量最多,分别占33 316 个分子功能Unigene 的45.80%和37.85%。
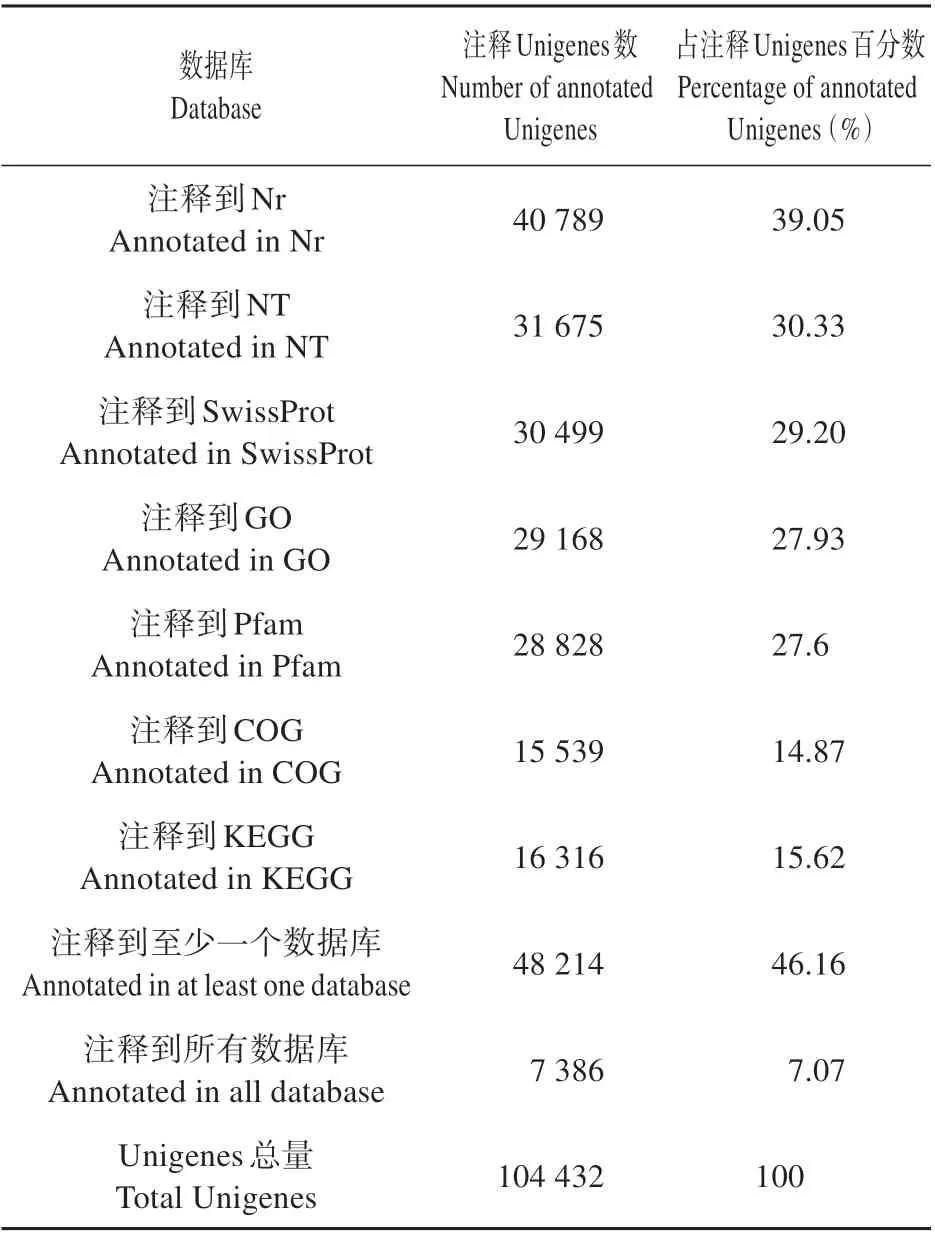
表2 泡桐Unigenes序列的功能注释Table 2 Functional annotation of P.tomentosa Unigenes
2.4 Unigene的COG功能分析
将104 432条泡桐Unigene与COG数据库进行比对,泡桐维管形成层中有15 539 个Unigenes 得到注释。从图2 可以看出,注释到COG 数据库中的Unigenes 共分为25 类。其中,注释数目最多的一类是2 351 个一般功能预测的Unigene,占总数的15.13%。接下来的一类是2 247个翻译后修饰、蛋白质折叠、分子伴侣注释的Unigene 和1 804 个翻译、核糖体结构和生物发生注释的Unigene,分别占总数的14.16%和11.61%。而核结构注释的Unigene(52 个)、细胞外结构注释的Unigene(43个)和细胞活性注释的Unigene(8 个)数目较少,占总数的比例分别为0.33%、0.28%和0.05%。另外590个未知功能注释的Unigene占总数的3.80%。
2.5 Unigene的KEGG通路注释
将104 432 条泡桐Unigene 采用Blastx 比对到KEGG 数据库,共有16 316 条Unigenes 得到注释,涉及到130条代谢通路中。Unigene注释序列最多的20个代谢途径列于表3。由表3可知,最有代表性的通路是“核糖体”(1 216 条)、“植物病原体相互作用”(646条),“碳代谢”(638条),其次是“氨基酸的生物合成”(559条)、“蛋白质在内质网中的加工”(533 条)和“剪接”(396 条)。在苯丙素生物合成(Phenylpropanoid biosynthesis)代谢途径中,共发现266 条Unigenes。其中与木质素合成相关的酶有苯丙氨酸氨裂解酶(Phenylalanine ammonia lyase,PAL)、肉桂酸-4-羟化酶(Cinnamic acid-4-hydroxylase,C4H)、肉桂醇脱氢酶(Cinnamyl alcohol dehydrogenase,CAD)、肉桂酰辅酶A 还原酶(Cinnamoyl-CoA reductase,CCR)和咖啡酸辅酶A-O-甲基转移酶(Caffeic acid coenzyme A-O-methyltransferase,CCoAOMT)。这些Unigenes 功能的注释为后续研究木材形成的分子机制起到极其重要的作用。
2.6 泡桐SSR特征分析
通过对泡桐Unigene的搜索,104 432个Unigenes 序列共检测出16 118 个SSR 位点(见图3),包含了9 001个单核苷酸、5 024个二核苷酸、2 048个三核苷酸、17 个四核苷酸、14 个五核苷酸和14 个六核苷酸重复序列。其中,单核苷酸中又以A/T重复基元为主(4 427 个)占全部单核苷酸重复的49.18%。AG/CT 类型在二核苷酸重复中最多,三核苷酸重复以AAG/CTT居多。
3 讨论
近年来,RNA-Seq 高通量测序技术已成为非模式生物遗传背景解析的重要手段,已经用于木本 植 物 杉 木(Cunninghamia lanceolata)[1]、柳 杉(Cryptomeria fortunei)[5]、柚木(Tectona grandis)[6]、
泡桐(Paulownia tomentosa)[8]和泡泡刺(Nitraria sphaerocarpa)[13]等转录组的研究。本研究利用Illumina 高通量测序技术对泡桐维管形成层及木质部区进行转录组测序,获得109 021 918 条clean reads,碱基Q30为94.30%,当Q30在80%以上就认为测序质量非常可靠[9]。对clean reads 片段进行拼接组装后共获得104 432 个Unigenes,平均长度为662 bp。其中N50 值为1 105 bp,说明本研究所得序列中长片段较多,组装效果较好,有利于进一步的研究分析。

表3 泡桐转录组Unigene的KEGG代谢途径分类Table 3 Classification of unigenes KEGG metabolic pathways in P.tomentosa transcriptome
将泡桐维管形成层及木质部区组装得到的Unigenes 与NR、GO、SwissProt、KEGG 和COG 等公共数据库进行序列比对,共有48 214(46.16%)个Unigenes成功获得注释,但仍有56 218(53.84%)个序列定位不清楚,未得到功能注释,这与许多木本植物杉木(Cunninghamia lanceolata)[1]、泡桐(Paulownia tomentosa)[8]、云南松(Pinus yunnanensis)[14]和穗花杉(Amentotaxus argotaenia)[15]均出现大量未获得注释Unigene 的结果类似。出现这一现象的原因之一可能是由于这些Unigene 片段较短或是本身为非编码RNA 序列[16]。研究表明,转录组中Unigene 的序列越长,获得注释信息的可能性就越高[9]。另外也有可能是这些大量未获得注释Unigene 是泡桐特有的新基因,这对后续开展泡桐维管形成层特异性分析提供重要信息。
RNA-Seq 高通量测序技术不仅使以较低的成本研究非模式生物基因组成为可能,同时也为SSR 位点的挖掘提供了大量序列信息[16]。曹玉婷等[5]在柳杉维管形成层转录组序列中鉴定获得3 772 个SSR 位点,占比最高的为三核苷酸重复SSR。王兴春等[17]利用MISA软件分析了连翘转录组,从Unigenes 中获得3 199 个SSR 位点。其中,占比最高的为单核苷酸重复SSR,其次是二核苷酸重复和三核苷酸重复。本研究利用MISA 软件分析了泡桐形成层及木质部区转录组,从长度为1 000 nt 以上的Unigenes 中获得16 118 个SSR 位点,其中9 001 个是单核苷酸重复的SSR,接下来是5 024 个二核苷酸重复和2 048 个是三核苷酸重复SSR,这与连翘[17]、杜仲[18]和莲雾[19]中发现的SSR 结果基本一致。这些结果将为泡桐SSR 标记开发提供重要序列信息,也为泡桐种质资源遗传多样性分析以及分子标记辅助育种等工作提供了重要基础。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
扬子江诗刊(2020年3期)2020-11-17
扬子江(2020年3期)2020-06-08
天津农业科学(2015年11期)2015-12-03
热带农业科学(2015年6期)2015-07-18
安徽农学通报(2014年7期)2014-04-29
吐鲁番(2014年2期)2014-02-28
吉林体育学院学报(2007年6期)2007-12-08
