
抗肿瘤天然拓扑异构酶抑制剂的研究进展
2021-03-17 00:08:54宋祖荣
中草药 2021年6期
戴 一,宋祖荣
抗肿瘤天然拓扑异构酶抑制剂的研究进展
戴 一1, 2,宋祖荣1
1. 安徽新华学院药学院,安徽 合肥 230088 2. 中国科学技术大学化学系,安徽 合肥 230026
拓扑异构酶(topoisomerase,topo)是解决DNA复制、转录等过程中出现拓扑问题的关键酶,是抗肿瘤药物作用的重要靶点之一。天然产物是topo抑制剂的重要来源,从天然产物中发现开发topo抑制剂是抗肿瘤药物研究的热点。以天然产物结构类型,分类综述了天然存在的具有topo抑制活性的天然产物及以它们为基础改造获得的衍生物,以期为抗肿瘤天然topo抑制剂的研发提供参考。
抗肿瘤;天然产物;拓扑异构酶抑制剂;结构改造;萜类;生物碱类;酚类;醌类
拓扑异构酶(topoisomerase,topo)广泛存在于生物体中,是细胞DNA复制或转录不可或缺的一类关键酶。由于DNA在复制过程中反向旋转会产生缠结、正负超螺旋等,为保证复制正常进行,必须依赖topo的参与进行DNA的切割、回旋、再连接,使DNA顺利解旋、复制、转录等[1]。根据topo诱导DNA断裂方式的不同,topo分为topo I和topo II 2种。人topo I为单体酶,主要催化DNA复制过程中单链的断裂和重新链接;topo II又称解旋酶,结构为二聚体,在三磷酸腺苷供能,Mg2+存在条件下,催化双链的断裂和链接[2]。相比于正常细胞,肿瘤细胞中topo的含量及活性均显著提高[3],因此topo是抗肿瘤药物优良的作用靶点,如喜树碱类、鬼臼毒素类、阿霉素等都是以topo为靶点,干扰DNA复制而发挥抗肿瘤作用的。
以topo为靶点的抑制剂是抗肿瘤药物研究的热点,这些topo抑制剂按照作用机制主要分为2大类:topo毒剂和topo催化抑制剂,前者通过形成DNA-topo I抑制剂三元复合物,达到抑制topo I催化作用的目的,如喜树碱类药物,或通过稳定topo II-DNA可裂解复合物而发挥作用,如鬼臼毒素类;后者主要通过嵌入DNA或直接作用于topo功能域而阻止topo与DNA的结合而发挥抗肿瘤作用,如阿霉素等[4]。通过文献报道发现天然产物是topo抑制剂的重要来源[5],多种天然来源的药物已广泛用于临床的肿瘤治疗,如羟喜树碱、拓扑替康、依托泊苷等。近年来基于topo为靶点的天然药物研究亦取得了积极进展,本文主要对2016年以来的成果进行综述,以期为新的topo抑制剂开发提供参考。
1 天然成分
1.1 萜类
天然产物中萜类化合物具有多种活性,尤其是抗肿瘤活性,受到广泛的关注,由于萜的骨架类型多样,为抗肿瘤药物的开发提供了丰富多样的先导化合物或候选药物[6]。萜类化合物作用的靶点多样,较成熟的如微管蛋白、topo等。近年来从植物获得的topo抑制剂主要为二倍半萜和二萜。如从Hedge的嫩芽中分离出线性的二倍半萜金合欢醇(1),从该植物根部分离出重排松香烷型二萜sahandinone(2)和4-脱氢沙维林醇(3),通过分子对接及分子动力学模拟研究,发现它们均能很好地与topo I结合,说明对topo I的抑制可能是其发挥抗肿瘤作用的机制[7]。
heteronemin(4)则是分自于海洋海绵sp.中的1个二倍半萜,通过消除topo II的主要酶活性,对topo II起到催化抑制的作用。该成分对前列腺癌LNcap和PC3细胞增殖具有显著的抑制活性,24 h结果显示半数抑制浓度(median inhibition concentration,IC50)值分别为1.4、2.7 μmol/L。在动物实验中也表现出显著的肿瘤抑制活性,而且对小鼠体质量几乎没有影响[8],体现了高效低毒的特点,具有进一步开发的潜力。从另一种海绵sp.中也获得二倍半萜12β-(3′β- hydroxybutanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al(5)和12β-(3′β-hydroxypentanoyloxy)- 20,24-dimethyl-24-oxo-scalara-16-en-25-al(6),这2个成分为scalarane型二倍半萜,另外还分离得到1个已知tetraprenyltoluquinol型化合物(7),这些产物通过抑制热休克蛋白(heat shock protein 90,Hsp90)和topo IIα的活性而发挥双重作用,促进凋亡[9]。可见,从海洋植物中寻找新型topo抑制剂具有一定潜力,尤其是寻找新的骨架成分。
真菌中亦常含有萜类成分,如从真菌壳囊孢属sp.中分离出1个杂萜类天然成分壳囊孢内酯C(8)。MTT实验发现其对人肺癌A549细胞、人结肠癌HCT-116细胞和人乳腺癌MCF-7细胞的IC50值在5.98~8.88 μmol/L,显示出了较好的抗增殖作用。合成该植物中另1个杂萜成分壳囊孢内酯A(9)的模型化合物四去氧壳囊孢内酯A(10)对这3种肿瘤增殖也表现出较强的活性,IC50为3.91~7.28 μmol/L,对其作用靶点研究发现在体外化合物8和10均具有类似喜树碱的topo I抑制活性,另外化合物10还对topo II表现出了抑制作用,效果与已知的topo II抑制剂ICRF-193相当[10]。对topo I和topo II同时抑制可以提高疗效,降低不良反应,属于多靶点抗肿瘤[11]。
2016年以来萜类topo抑制剂的化学结构见图1。
1.2 生物碱类
生物碱类成分也是天然产物中结构类型多样的一大类成分,具有的生物活性促使很多天然成分已直接应用于临床,如作用于M受体的阿托品,作用于微管蛋白的长春新碱以及众所熟知的topo I抑制剂羟喜树碱[12]。近年来海洋植物成分研究是天然药物研究的热点。奇西卡马碱A(11)和它的类似物16,17-去氢奇西卡马碱A(12)是从南极深海海绵Kirkpatrick中分离的2个吡咯亚胺醌类生物碱,通过分子对接发现这2个化合物对topo I和topo II具有较好的结合,表现出双重抑制潜力[13],为抗癌药物先导化合物的开发提供了一个新的选择。另外从温带海洋一种海绵Samaai & Kelly中分离出来5个吡咯亚胺醌类生物碱makaluvamine Q(13)、makaluvamine A(14)、makaluvamine I(15)、tsitsikammamine B(18)、14-bromo-7,8-dehydro-3-dihydro-discorhabdin C(19)、2个吡咯邻醌类makaluvamine O(16)和makaluvone(17)。所有的化合物对topo I均显示了抑制作用及DNA嵌入功能,其中化合物16、17活性最强,对宫颈癌HeLa细胞增殖具有中等抑制作用[14]。此外,2019年从地中海萍蓬草(L.) Sm. ssp.(Timm) E. O. Beal中分离出1个活性天然成分6,6′-二羟基硫双萍蓬草碱(20),该结构为含硫的二聚倍半萜形成的萍蓬草生物碱,是topo II毒剂,通过与topo II共价键结合发挥抑制作用,而且体现出一定选择性,抑制topo IIα的作用强于topo IIβ[15]。海洋植物受到的环境胁迫不同于陆生植物,从海洋植物中常可获得结构新颖的成分,再次说明海洋植物对topo抑制剂的开发具有巨大潜力。
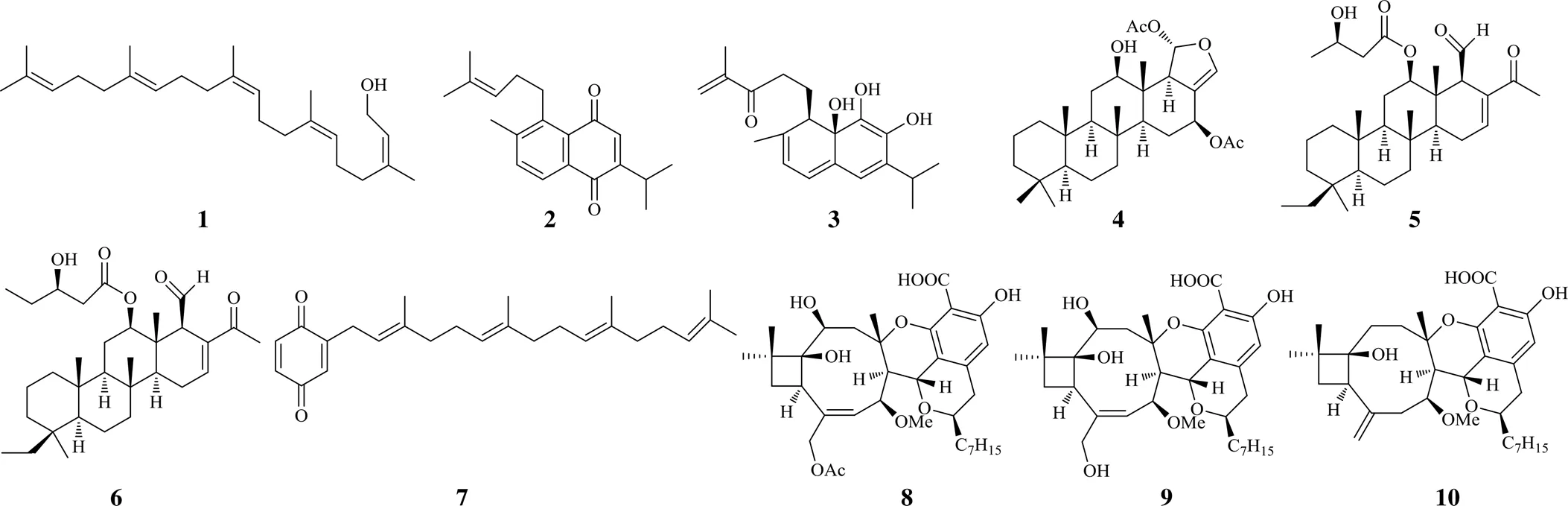
图1 2016年以来萜类topo抑制剂的化学结构
对陆生植物,近年来主要从抗炎植物两面针(Roxb.) DC.中获得1个生物碱氯化两面针碱(21),该生物碱通过抑制topo I而促进白细胞介素-10的产生,抑制了炎症反应,在体内、体外表现出显著的抗炎作用[16]。近年来的研究发现,炎症和癌症的发生发展密切相关[17],而该成分又是通过抑制topo来发挥作用的,值得进一步深入研究。
2016年以来生物碱类topo抑制剂的化学结构见图2。
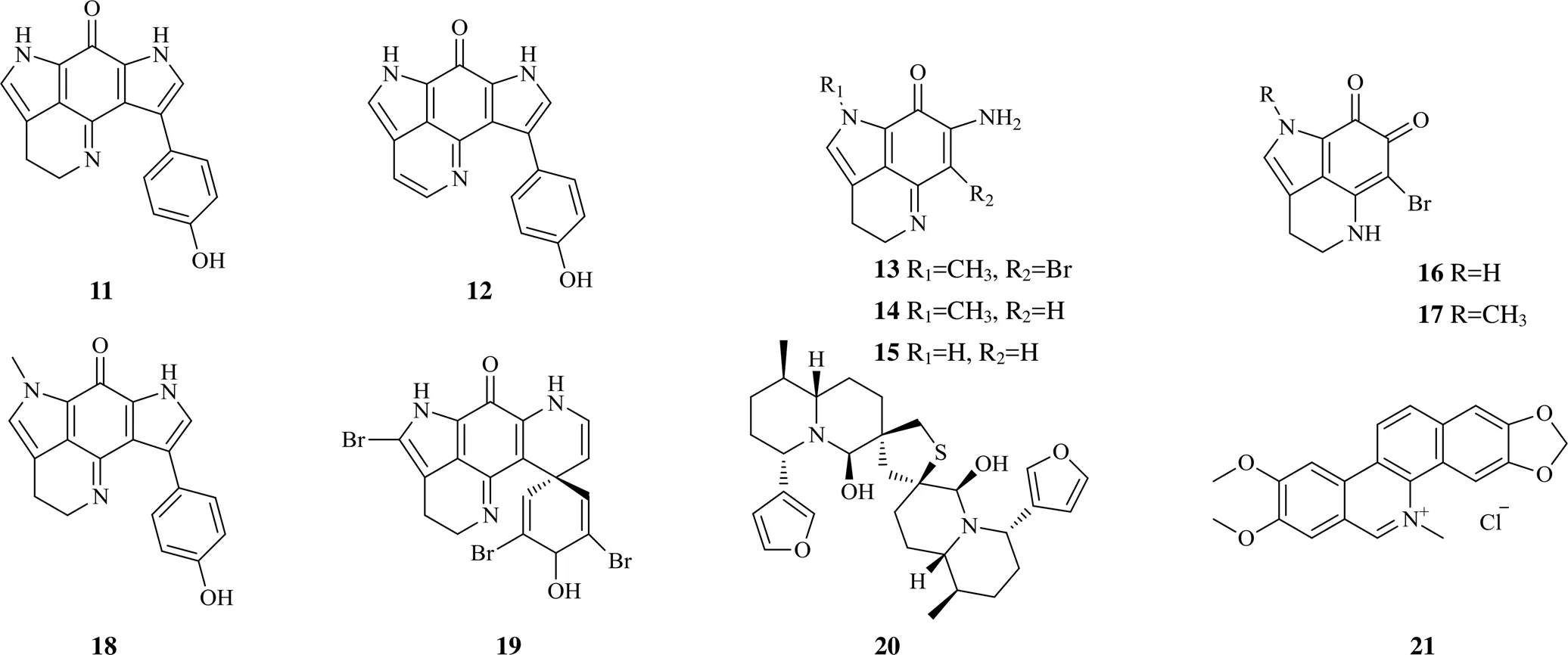
图2 2016年以来生物碱类topo抑制剂的化学结构
1.3 酚类
酚类化合物广泛存在于自然界,是个庞大的复合家族,包括黄酮类和非黄酮类的酚性物质等,近年来在抗肿瘤方面的研究备受关注[18]。乔松酮(22)即为天然黄酮类成分,可以结合topo I和DNA的表面形成四元复合物,而对topo I产生别构抑制[19]。对来自珊瑚上的真菌和植物进行分离获得(−)-表没食子酰儿茶精-3--反式-对-香豆酸酯(23)、(−)-表没食子酰儿茶精-3--顺式-对-香豆酸酯(24)、(−)-表棓儿茶素(25)、槲皮素(26)、(−)-没食子酰儿茶素(27)、交链孢毒素I(28)、6--stemphytriol(29)和多黏菌素C(30)。这些成分在5~100 μmol/L显示出抑制topo I诱导的DNA超螺旋松弛活性,尤其是,黄酮类成分比其他成分的抑制topo I能力更强。这些新发现的topo I抑制剂显示了结构的多样性,是抗癌先导化合物开发的一个新选择。考察更低浓度时这些成分对topo的抑制活性时发现,4个化合物23~26显示在25 μmol/L有效,其中化合物23、24显示在更低的浓度(5 μmol/L)也有效。化合物23、24相比于表没食子儿茶素没食子酸酯(epigallocatechin gallate,EGCG,10 μmol/L)显示出了更高的抑制活性[20]。
3个五角型多酚amexanthomycins A~C(31~33)是从地中海拟无枝菌酸菌S699ΔrifA中分离出来的,该菌株敲除了与利福霉素合成相关的聚酮合成酶基因。化合物31~33显示了对DNA topo的抑制作用。采用DNA松弛实验研究化合物31~33对Topo IIα的抑制活性,500 μmol/L时显示有效。剂量相关的活性测定显示化合物31和32相比于化合物33对topo IIα抑制活性更强。这些结果说明在amexanthomycins上取代基去氧糖对topo的抑制作用有重要影响,对于开发新型topo抑制剂具有参考价值[21]。
2016年以来酚类topo抑制剂的化学结构见图3。
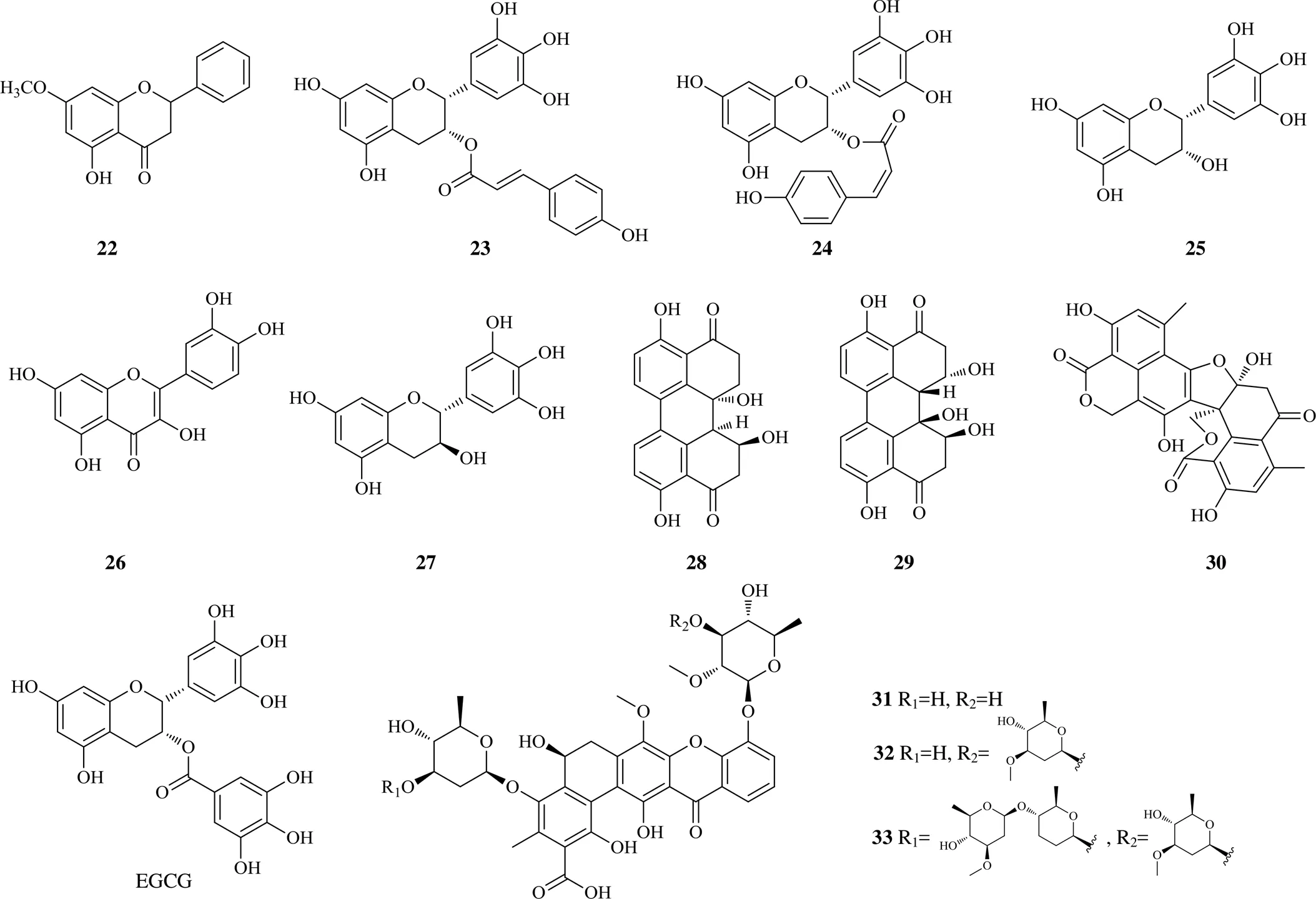
图3 2016年以来酚类topo抑制剂的化学结构
1.4 醌类
醌类化合物是中药中一类具有醌式结构的化学成分,临床上使用的topo抑制剂阿霉素属于该类。同阿霉素来源类似,该类成分多来源于微生物。pyxidicycline A(34)和pyxidicycline B(35)是利用自身抗性导向的基因组挖掘策略从匣状粘球菌An d48分离出来的2个聚酮型天然产物,对人结肠癌HCT-116细胞最低抑菌浓度(minimum inhibitory concentration,MIC)分别为0.06、0.03 μg/mL,对人topo I的IC50为0.4~1.6、0.05~0.2 μg/mL,与喜树碱相当[22]。另一类聚酮天然产物为jadomycin,来自于土壤委内瑞拉链霉菌ISP5230,jadomycin B(36)、jadomycin S(37)和jadomycin F(38)对药物敏感型人乳腺癌MDA-MB-231细胞和紫杉醇耐药性MDA-MB-231细胞增殖表现出同样的抑制作用,进一步的研究发现它们能抑制topo IIα和topo IIβ的基因和蛋白的表达,同时也是topo II的催化抑制剂,在某些情况下也表现为topo II的毒剂。化合物36和38还表现出对topo IIβ更高的选择性[23]。
GE-1(39)和GE-2(40)来自于子囊菌,具有抗肿瘤活性,化合物40是39的氧化型。像依托泊苷一样,在1个专一的位点诱导DNA断裂,化合物40显著提高DNA断裂程度。与其他醌类topo II毒剂类似,化合物40通过共价键与topo II结合。与化合物40相比,化合物39要想达到相似的效果需要较高的浓度才能实现,可见氧化态结构能使活性得到提高[24]。从现有的成果可见微生物来源的topo抑制剂表现出结构上的共性,多为醌类,是天然topo抑制剂的另一种展现。
2016年以来醌类topo抑制剂的化学结构见图4。
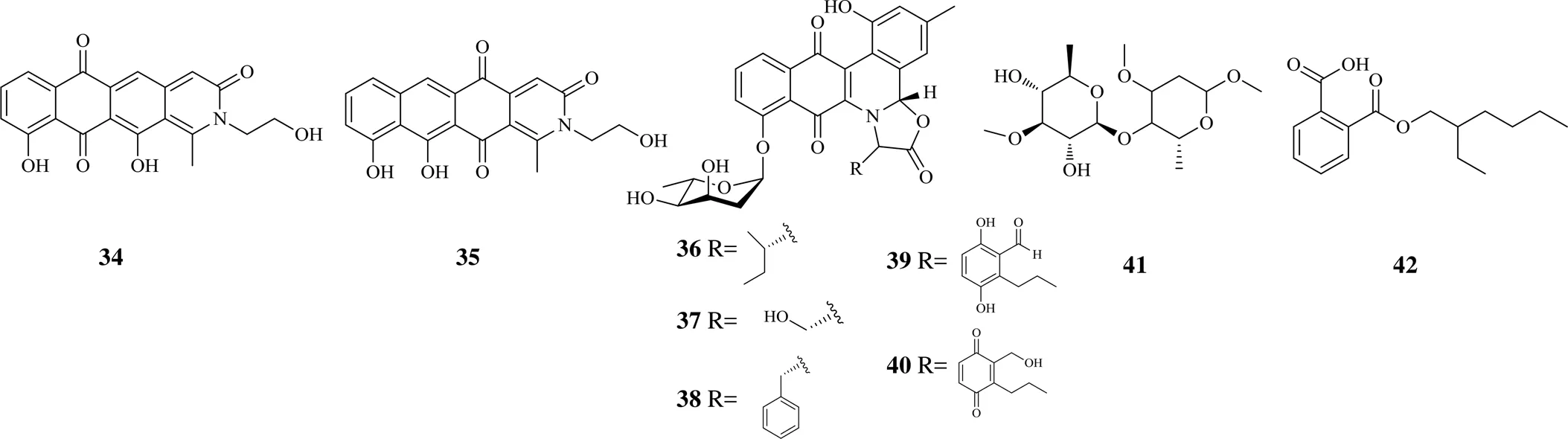
图4 2016年以来醌类topo抑制剂的化学结构
1.5 其他
其他具有topo抑制活性的天然成分类型较少,近年来主要发现1个苷类成分和1个芳香酸酯。methyl β-lilacinobioside(41)为从沙特阿拉伯南部植物巨棱阁N. E. Br.分离出来的1个苷类新成分,对MCF-7细胞的增殖具有较强的抑制作用,通过分子对接实验发现其与topo II氢键和离子键结合,与topo II的亲和力达−36.975 kJ/mol,说明对topo II的抑制应是其发挥抗肿瘤活性的重要因素[25]。另1个为芳香酸酯即邻苯二甲酸单(2-乙基)己酯(42),来自于海洋菌株sp. VITJS4,对人肝癌HepG2细胞增殖有显著的抑制活性,也通过分子对接及分子动力学模拟研究发现,其作用的靶点是topo IIα的ATPase结构域[26]。
2 天然成分结构改造
2.1 生物碱类成分改造
从生物碱改造获得先导化合物持续受到关注[27],近年来对生物碱类成分的结构改造以喜树碱衍生物7-乙基-10-羟基喜树碱(7-ethyl-10-hydroxycamptothecin,SN-38)修饰较多。SN-38通过抑制topo I阻滞DNA的复制而发挥抗肿瘤作用[28]。伊立替康即为SN-38的水溶性前药,但体外活性比SN-38低100~1000倍[29]。体内仅有使用剂量的2%~8%能转化成SN-38[30]。因此,SN-38依然受到很大的关注。对SN-38改造的位点主要为9位和10位。9位的改造主要是引入不同的氨甲基,获得化合物43a~43d,这些氨甲基衍生物对人白血病HL-60细胞、MCF-7和MDA-MB-231细胞、结直肠腺癌Caco-2和HT-29细胞及A549细胞的增殖抑制作用与SN-38相当,和拓扑替康一样,通过共价键结合天然DNA八聚体d(GCGATCGC)2duplex的边缘GC碱基产生topo I抑制作用,即使在10 ℃也能在一定时间后共价键结合寡聚核苷酸[31];对于10位的修饰,分别以醚键或酯键在10位引入硝基咪唑基团 (1-甲基-2-硝基-1-咪唑-5-基) 甲醇,获得化合物44和45,这2个化合物为低氧激活型SN-38的前药。化合物45的低氧选择性及细胞毒性均弱于化合物44,在低氧条件下化合物44对人大细胞肺癌H460细胞增殖的抑制活性是evofosfamide的10倍以上,与SN-38相当[32]。另1个10位修饰的化合物46,为SN-38与氨基酸以酯键相连,该化合物对A549细胞和人肺癌SPC-A-1细胞、人白血病K562细胞、MCF-7细胞、人小细胞肺癌NCI-H446细胞、人卵巢癌SK-OV-3细胞、人肝癌SMMC-7721细胞、人结肠腺癌SW1116细胞和胶质瘤U251细胞9种人肿瘤细胞增殖的IC50值为0.000 9~2.567 1 μmol/L,均低于伊立替康或SN-38,表现出更强的肿瘤抑制作用,在稳定topo-1-DNA复合物方面比伊立替康及SN-38更强[33]。
2016年以来SN-38衍生物的化学结构见图5。
除了对SN-38修饰外,2018年还报道了在喜树碱或羟喜树碱的7位引入醛基,通过亚胺和沙丙素A进行组合获得杂合体47a~47c,这3个化合物具有topo I抑制及HDAC抑制双重作用,分子模拟证实了这种功能,该杂合体对人肺癌NCI-H460细胞、人胰腺癌CAPAN1细胞、表皮癌A431细胞、HeLa细胞、人结肠癌HT29细胞、前列腺癌DU45细胞、HepG2细胞、人卵巢癌A2780细胞及耐阿霉素A2780-Dox细胞等多种肿瘤细胞均具有显著的抑制活性,IC50为0.05~0.8 μmol/L,而对照沙丙素A的IC50值约为0.9 μmol/L,伊立替康则大于1.5 μmol/L[34]。可见杂合体发挥多靶点功效,实现了协同作用。
2016年以来喜树碱和羟喜树碱衍生物的化学结构见图6。
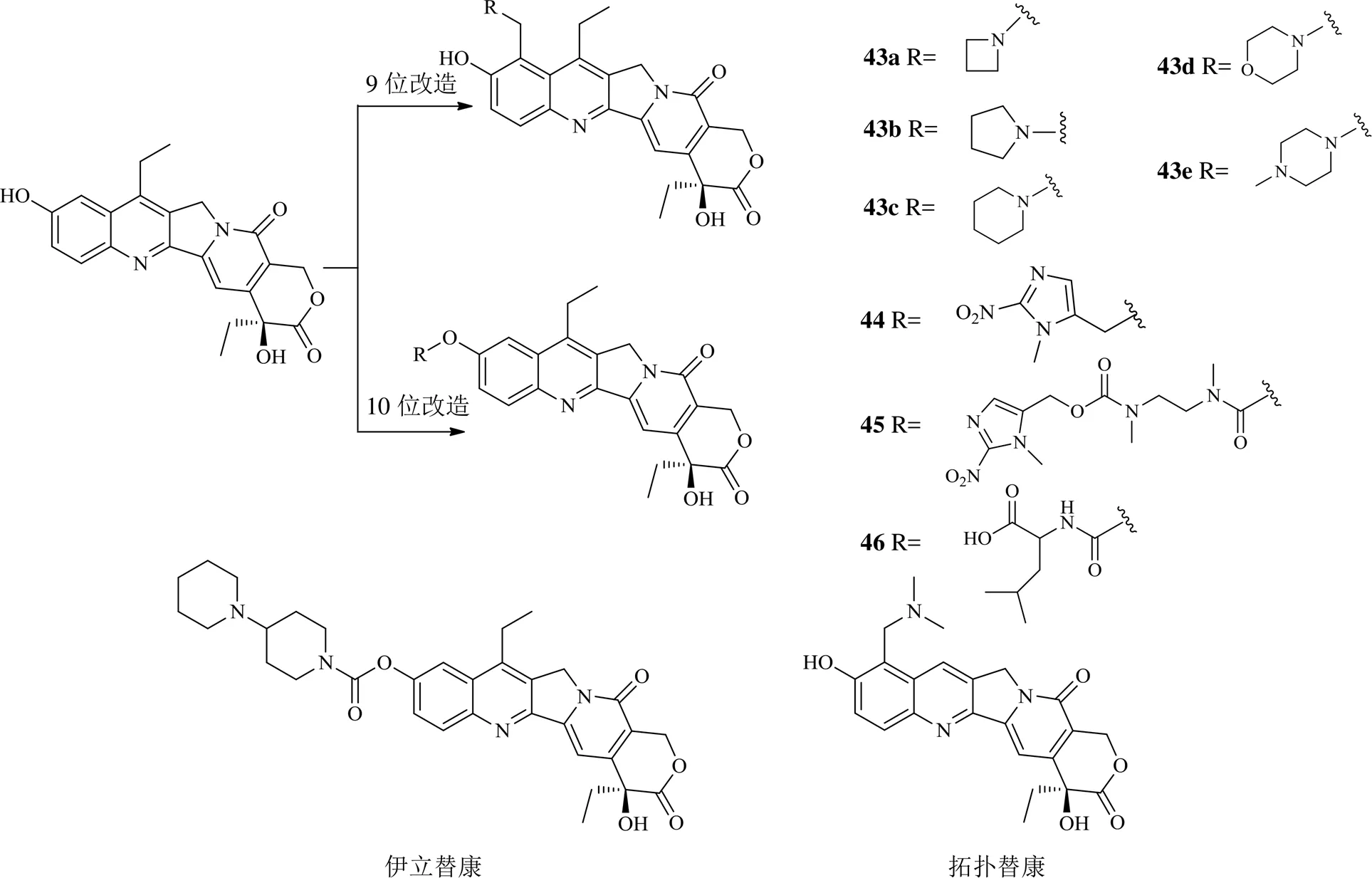
图5 2016年以来SN-38衍生物的化学结构
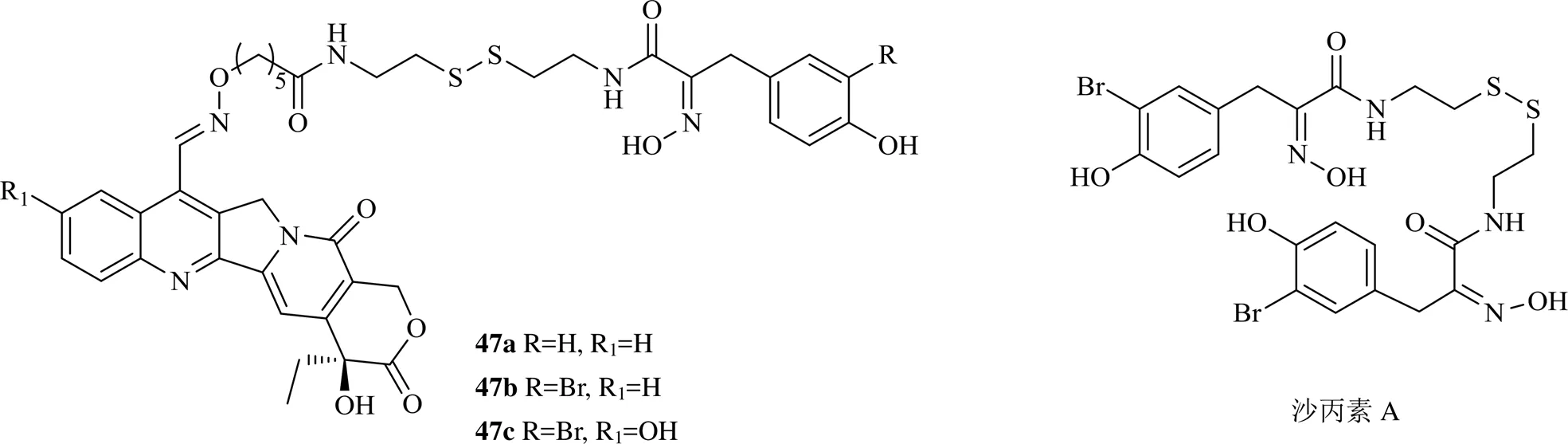
图6 2016年以来喜树碱和羟喜树碱衍生物的化学结构
此外,通过点击化学的合成手段在7-甲基-10-羟基高喜树碱10位引入芳烃、饱和烃或糖等合成化合物48a~48u,除了48h、48r和48u,这些衍生物对A549细胞、人乳腺癌MDA-MB-435细胞及人结肠癌HCT116细胞表现出较强的抗增殖作用,尤其对A549细胞,有12个化合物IC50值小于1 μmol/L,对其中4个化合物48e、48j、48o和48t进行topo I毒性测定,化合物48j的活性高于喜树碱,其他3个化合物的活性类似于喜树碱或略低于喜树碱[35]。
2016年以来7-甲基-10-羟基喜树碱衍生物的化学结构见图7。
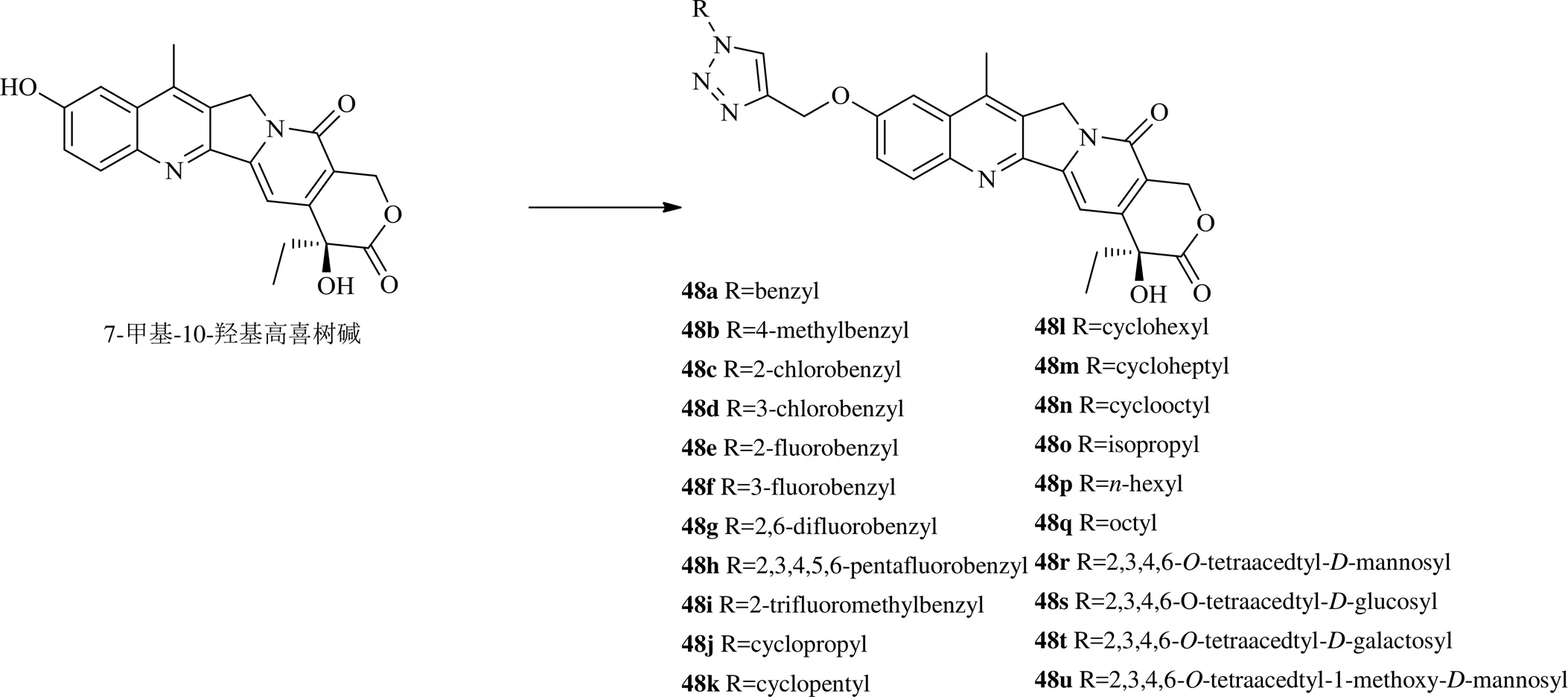
图7 2016年以来7-甲基-10-羟基喜树碱衍生物的化学结构
近年来另1个受关注的生物碱为来自于苦豆子L的喹诺里西啶类生物碱槐定碱,用于治疗滋养细胞肿瘤。对其改造的方式有:一是D环打开与磷酰胺氮芥通过酰胺键而形成杂合体,获得化合物49a~49e。因topo I是槐定碱的作用靶点,氮芥也能作用于DNA,通过分子对接实验发现49a~49e通过与topo I活性口袋中的氨基酸残基及DNA碱基双重作用与DNA-topo I共价复合物进行很好结合,具有topo I抑制作用[36];第2种改造是D环打开后羧基还原为羟基,氨基烷基化得化合物50,该化合物相对于母体化合物,抗肿瘤谱及活性均有了显著的提高,其机制是通过稳定DNA-topo I复合物而抑制topo I的活性,通过促进topo I介导的DNA单链和双链的断裂而诱导线粒体途径的凋亡[37]。
2016年以来槐定碱衍生物的化学结构见图8。
此外2019年还报道了1个结构类似于喜树碱的天然产物luotonin A,在此结构母体1~4不同位置引入氨基获得化合物51a~51d,化合物51d水溶性太差,其他3个均进行了MTT实验,对人白血病HL60细胞,化合物51b的抑制作用显著强于母体化合物luotonin A,而化合物51a、51c活性与luotonin A相当;对于人结肠癌SW480细胞,化合物51a和51b的活性均强于母体luotonin A,化合物51c则与luotonin A相当。对于DNA topo I的抑制作用实验发现化合物51a、51b、51d均表现出比喜树碱更强的抑制活性,而化合物51c则没有抑制作用[38]。
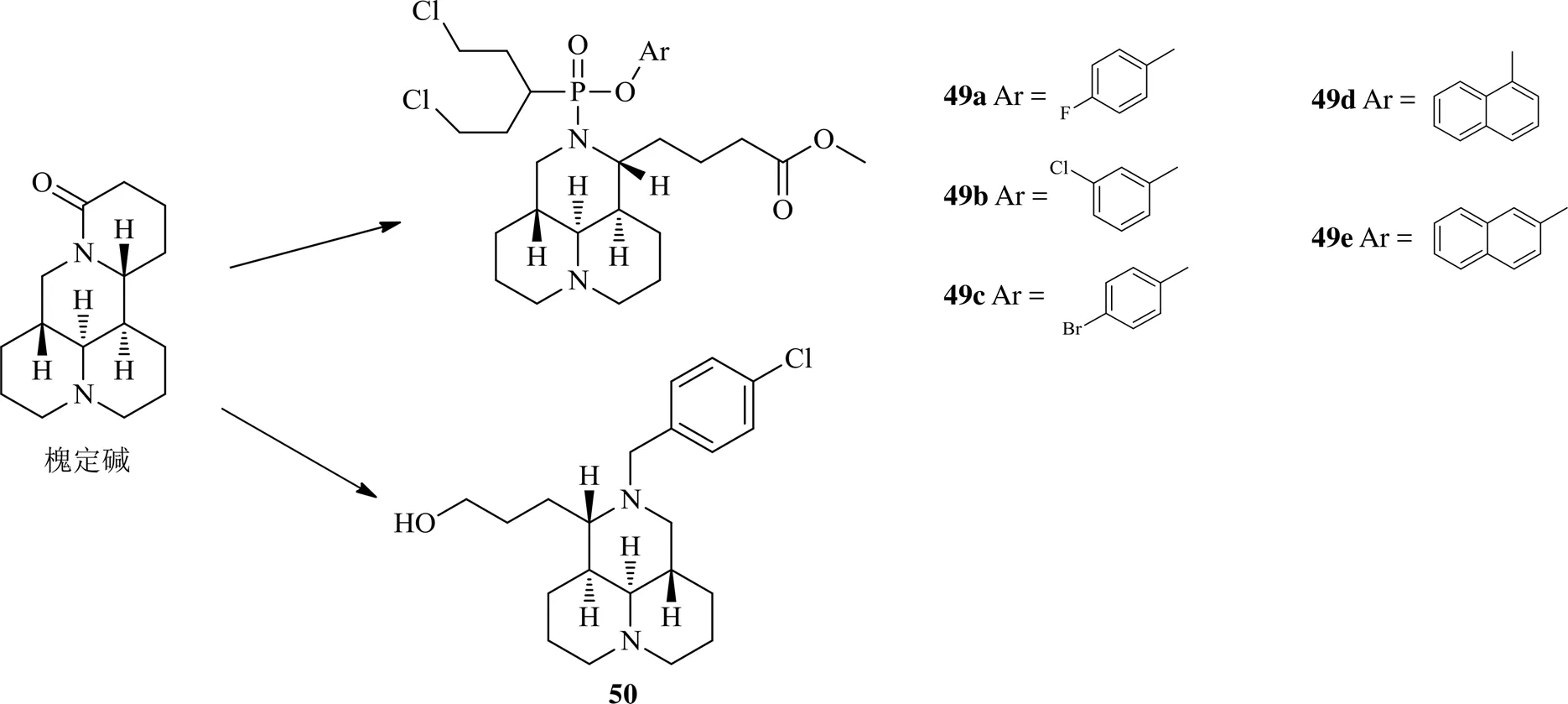
图8 2016年来槐定碱衍生物的化学结构
玫瑰树碱是来自于热带一种常绿小树古城玫瑰树Labill的生物碱,由于其独特的平面四环结构成为了抗癌药物研发热点,多个衍生物已进入临床实验[39-40]。鉴于该化合物对topo II的抑制作用,进行11位的取代修饰,获得3个化合物52~54,且均表现出对topo II有较好地抑制作用。化合物52~54在DNA松弛实验中100 μmol/L对topo II显示出良好的抑制活性,尤其是11位连有α,β-不饱和羰基的化合物53和9位连有亚胺,11位为甲酰的化合物54具有更大的潜力,体现着一种新的开发角度,为今后该类化合物活性的提高提供了参考[41]。
2016年以来luotonin A和玫瑰树碱衍生物的化学结构见图9。
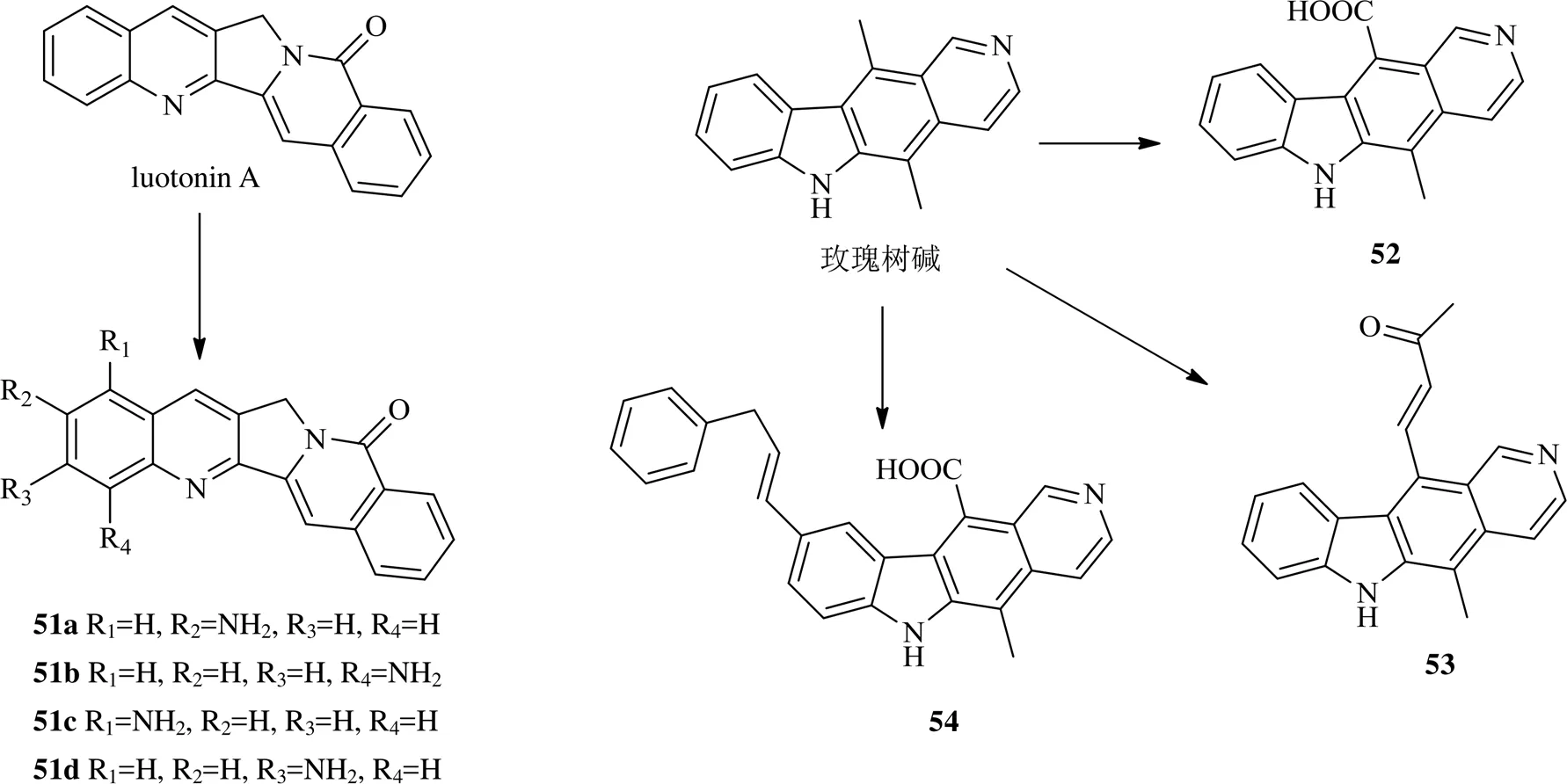
图9 2016年以来luotoninA和玫瑰树碱衍生物的化学结构
吴茱萸碱是来自吴茱萸的1个吲哚吡嗪喹唑酮生物碱,为topo I、II双抑制剂,通过稳定topo I、II-DNA复合物导致DNA断裂而诱导细胞凋亡。对吴茱萸碱的结构分析,通过骨架迁跃合成了化合物55a~55d。这些化合物在100 μmol/L时可以抑制topo I介导的DNA超螺旋松弛,但对topo II没有作用,进一步的研究发现同喜树碱一样,化合物55b作为topo I的毒物而起到作用,另外化合物55b也可以浓度相关性地抑制微管蛋白的聚合,IC50为26.3 μmol/L,表现出对topo I和微管蛋白具有明显的选择性[42]。
2016年以来吴茱萸碱衍生物的化学结构见图10。
2.2 木脂素类衍生物
木脂素类成分修饰主要围绕鬼臼毒素展开[43],且常以4位修饰为主,如以鬼臼毒素为起始物在其4位引入4β羧基三唑,合成出了化合物56a~56i和57a~57k等多个鬼臼毒素的衍生物,这些衍生物显示出了很高抗增殖作用,对HeLa细胞、MCF-7细胞、人前列腺癌DU-145细胞、A549细胞、HepG2细胞和HT-29细胞等增殖的IC50值在1~10 μmol/L,尤其是化合物56b、56g和56i,IC50值低于1 μmol/L,比依托泊苷还强。topo介导的DNA松弛实验显示这些代表性衍生物56b、56g、56i均能有效抑制topo II的活性,与依托泊苷相当[44]。另一种鬼臼毒素的4位引入4β--乙酰胺获得化合物58a~58t等20个鬼臼毒素衍生物,通过对4个肿瘤细胞(人食管癌EC-9706细胞、HeLa细胞、人膀胱癌T-24细胞和H460细胞)和1个正常细胞(人正常皮肤HaCaT细胞)的MTT实验发现化合物58f的活性最好,对这些肿瘤细胞增殖的IC50值在1.2~22.8 μmol/L,低于对照依托泊苷(8.4~78.2 μmol/L)。化合物58e对HeLa细胞、T-24细胞有着更高的细胞毒性,鉴于鬼臼毒素作用topo,对所得衍生物以化合物58e为代表进行了分子对接实验显示化合物58e可以与topo IIα很好的结合,说明还保留鬼臼毒素作用的靶点[45]。
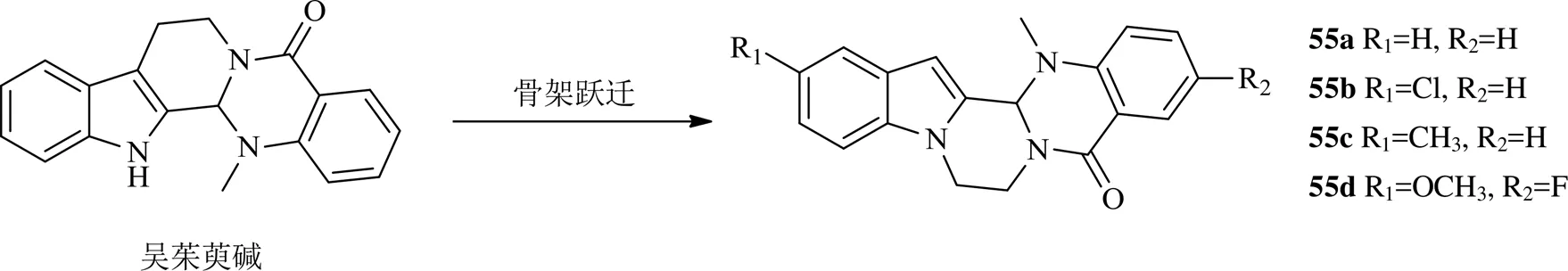
图10 2016年以来吴茱萸碱衍生物的化学结构
2016年以来鬼臼毒素类衍生物的化学结构见图11。
叶下珠素类是另一类含内酯类的芳基萘类木脂素,主要来源于植物叶下珠L.,该类成分主要是以山荷叶素为苷元与多种糖基形成的苷,在结构上与鬼臼毒素类相似,具有良好的抗肿瘤作用。鉴于鬼臼毒素类化合物作用于topo II,改造山荷叶素,寻找新的topo抑制剂,获得以山荷叶素为苷元的芳基萘类木脂素化合物59~61,结构类似于叶下珠素,对HT-29细胞的IC50值在18~140 nmol/L,对topo II的抑制作用比其母体化合物山荷叶素强,但弱于依托泊苷[46]。
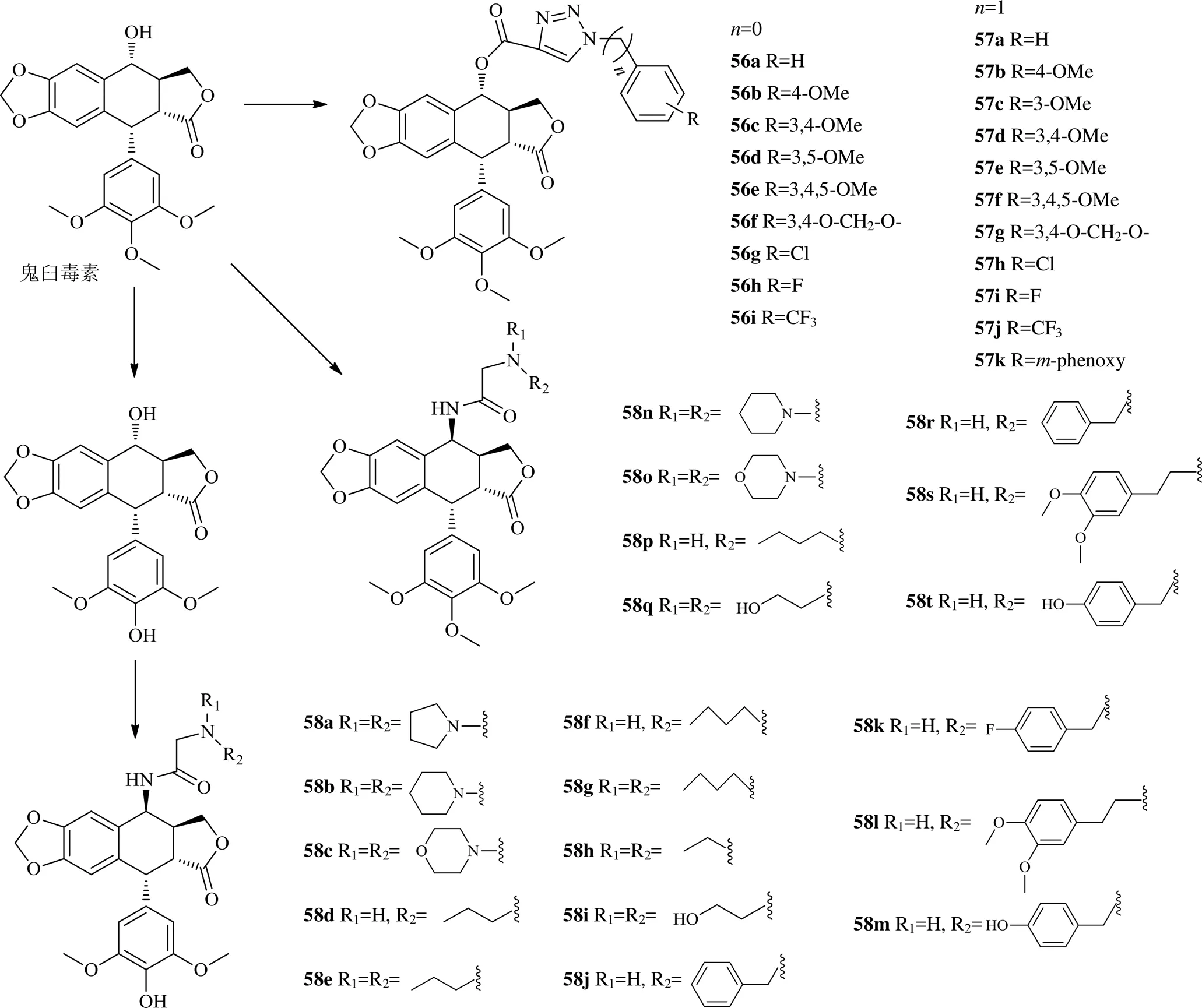
图11 2016年以来鬼臼毒素类衍生物的化学结构
2016年以来山荷叶素素类衍生物的化学结构见图12。
2.3 黄酮及萜类衍生物
黄酮类是天然产物中最为常见的成分之一,包括黄酮、异黄酮、查耳酮等,其中噢哢亦属于黄酮类,且具有topo抑制作用[47]。对噢哢进行骨架跃迁可以设计出合成2-芳基亚甲基咪唑[1,2-a]吡啶酮类化合物,如化合物62a~62j(图13)。这些化合物相比于依托泊苷,对topo IIα-介导的DNA断开抑制能力更强,在相同的取代条件下,化合物62c、62f相比于母体橙酮(C1和C2),对topo IIα的抑制作用更强。另外这些化合物对MCF-7细胞的抑制浓度在nmol/L水平[48]。说明了骨架跃迁设计思路对于噢哢类化合物开发是一个较佳的选择。
对黄酮及异黄酮同样可以采取骨架跃迁设计策略,如由黄酮设计出2-芳基-4-吡啶并[1,2-a]嘧啶- 4-酮类化合物63a~63g,由异黄酮设计出3-芳基- 4-吡啶并[1,2-a]嘧啶-4-酮类化合物64a~64i(图14)。这些化合物及其母体C1~C4、C6~C7对topo IIα抑制作用均比依托泊苷强,通过选择性抑制topo IIα的催化作用,而不干扰DNA大沟结构域处topo I与DNA的相互作用。对人胚肾HEK-293细胞IC50值在1.5~4.0 μmol/L。与母体化合物相比,通过骨架跃迁设计的化合物保留了相似功能,有的还提高了对topo IIα的抑制作用[49],为众多的黄酮类化合物的开发提供了一种新的思路。
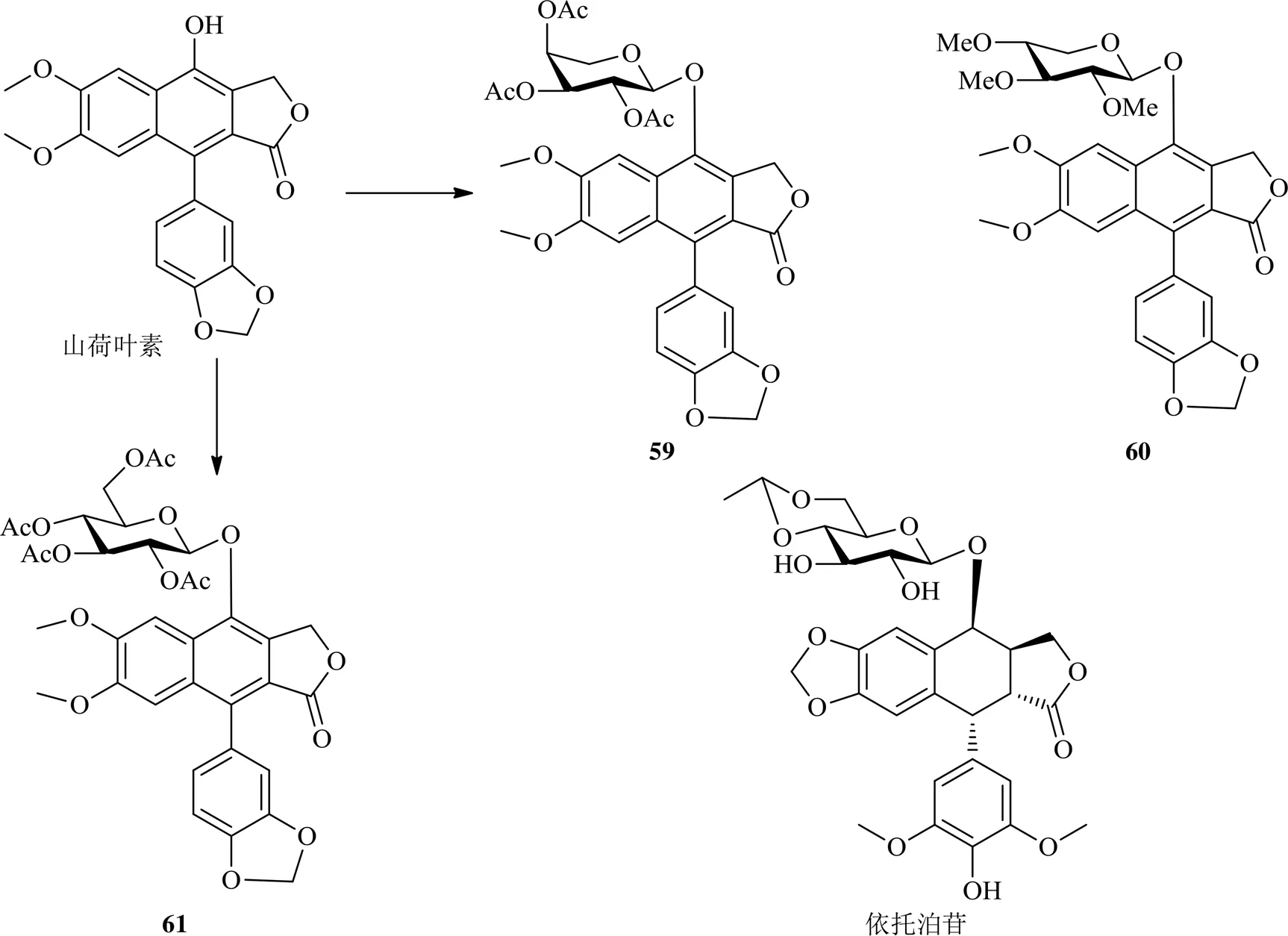
图12 2016年以来山荷叶素素类衍生物的化学结构
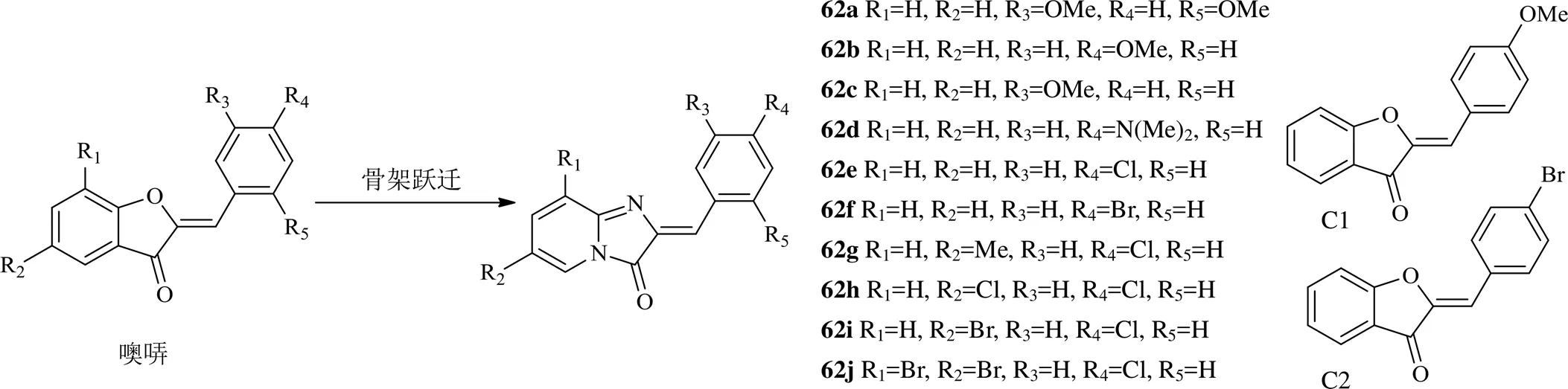
图13 2016年以来噢哢类的结构改造
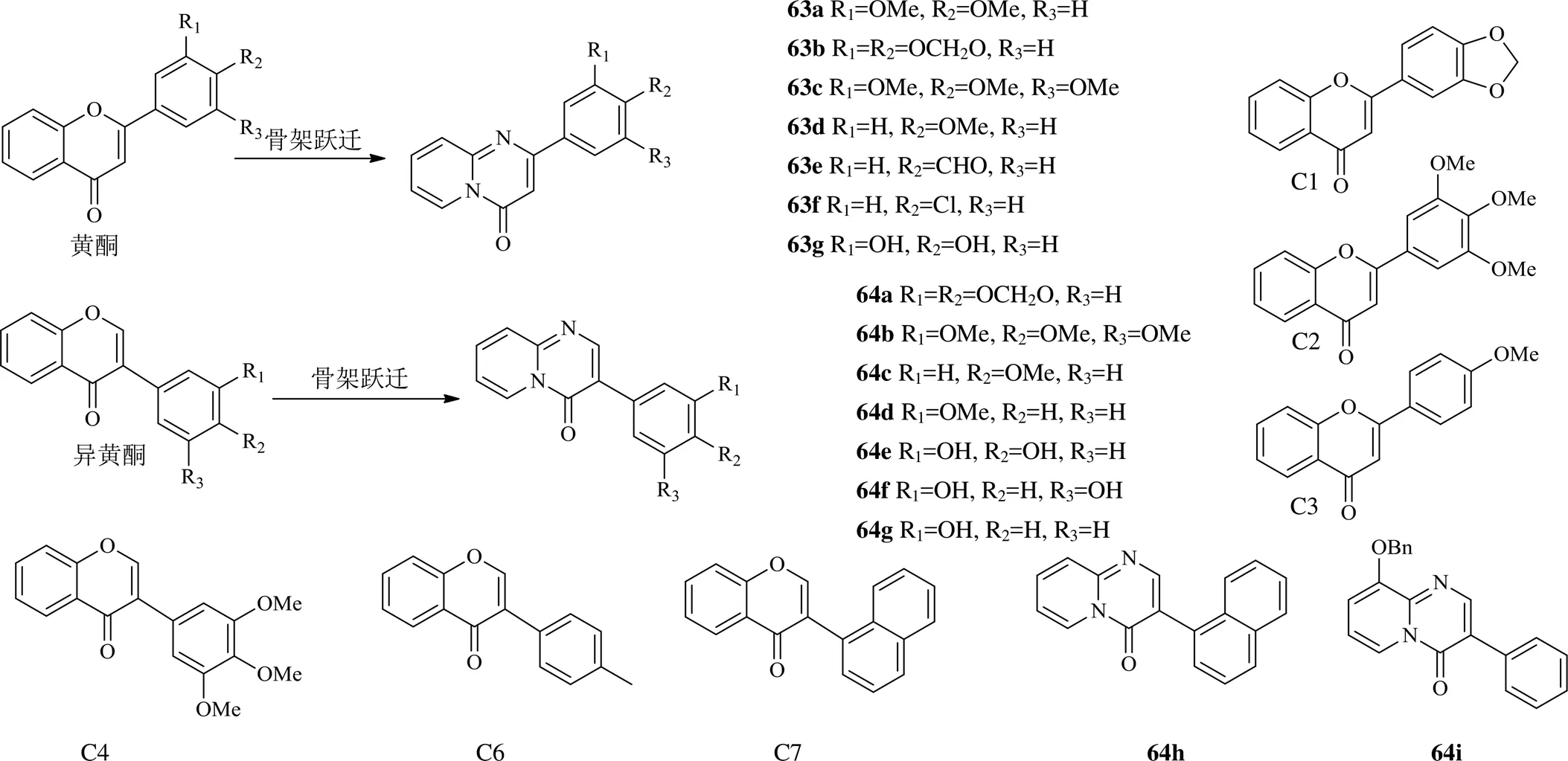
图14 2016年以来黄酮类的结构改造
萜类成分是抗肿瘤天然产物中的佼佼者,如紫杉醇等,是抗肿瘤药物开发中良好先导化合物来源之一[50-51]。反式-去氢巴豆宁是从巴西亚马逊巴豆Benth的茎皮中分离出来的1个克罗烷型二萜,以其为母体化合物,在酮羰基上经亚胺修饰即可获得化合物65~69(图15)。获得的化合物66~69对腹水瘤Ehrlich细胞的抑制作用明显强于母体化合物。同时化合物65~67、69对K562细胞具有一定的抑制作用,IC50为7.85~40.72μmol/L。进一步的机制研究发现,除了化合物66之外,这些化合物对topo I均有较强的抑制作用,尤其是化合物67作用最强[52]。可见,通过对天然产物进行修饰,即使是初步的修饰,也可能获得较大的收益。
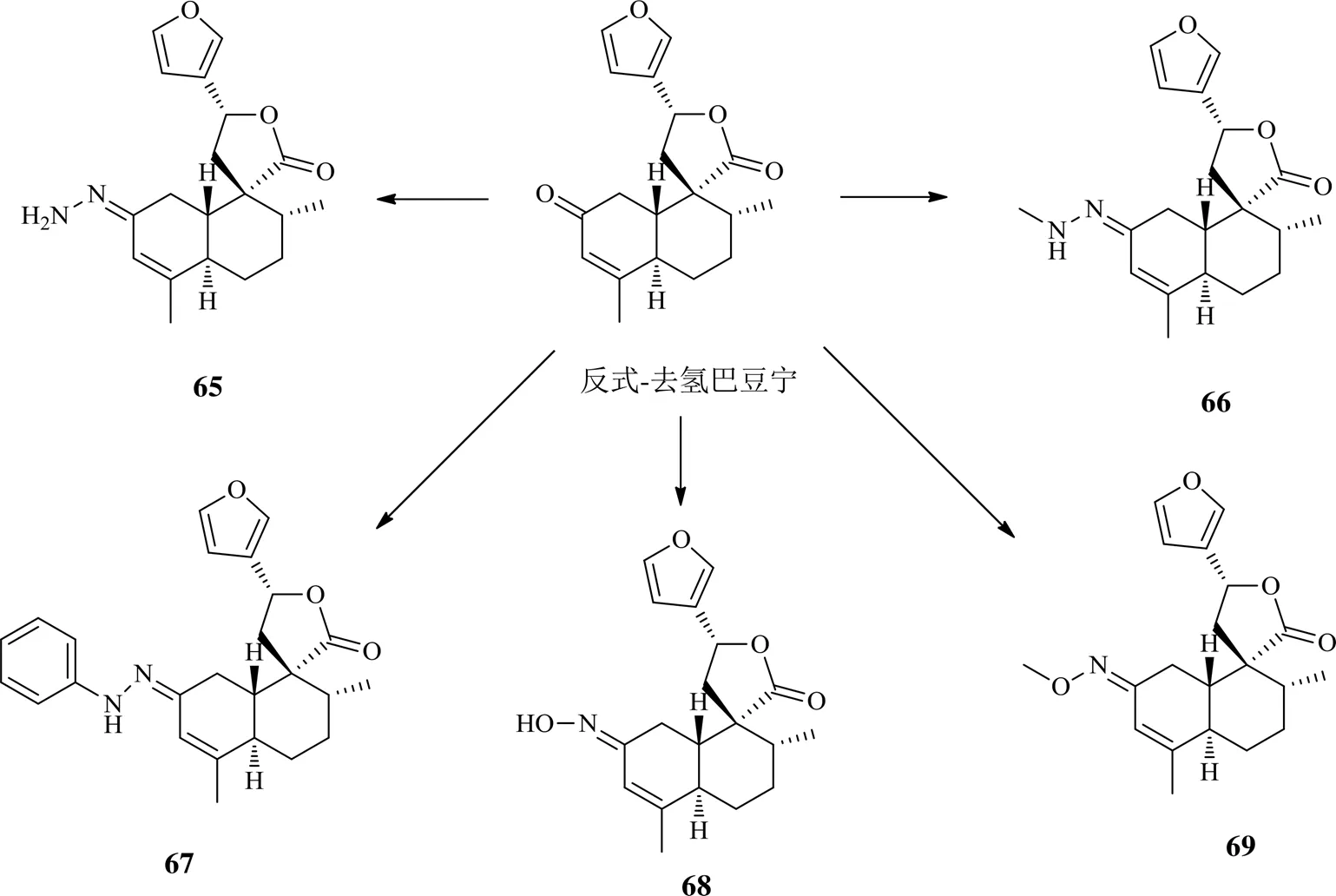
图15 2016年以来反式-去氢巴豆宁衍生物的化学结构
3 结语
天然产物是抗肿瘤药物的重要来源之一,分布在不同科属植物中,成分类型多样,涉及几乎所有成分类型[53]。天然产物抗肿瘤作用的靶点各异,DNA topo是其作用靶点之一,受到了广泛的关注。目前已发现许多种天然topo抑制剂,如生物碱类、木脂素类、黄酮类等,在新药的开发中提供了众多重要价值的先导化合物,如以羟喜树碱为基础开发出的拓扑替康、伊立替康;以鬼臼毒素为基础开发出的依托泊苷,它们已成为临床抗肿瘤的重要药物。近年来,天然topo抑制剂亦取得了很多成果。然而,纵观天然topo抑制剂,可以发现,成分类型比较集中,多以生物碱为主,其他结构类型的天然topo抑制剂较少,而且最近发现的很多成分来自于海洋植物,说明海洋是未来发现天然topo抑制剂的一个主要来源。由于天然产物本身存在的局限,如水溶性较差,活性不强,毒性太大等,以天然成分为基础,进行改造、修饰、转化,是一种行之有效的药物开发策略,也是今后药物开发的重点,近年来的研究成果也充分体现了这点,因此以天然产物为基础,进行深层次结构开发是topo抑制剂开发的一个重要方向。
利益冲突 所有作者均声明不存在利益冲突
[1] Wang J C. Cellular roles of DNA topoisomerases: A molecular perspective [J]., 2002, 3(6): 430-440.
[2] Nitiss K C, Nitiss J L, Hanakahi L A. DNA damage by an essential enzyme: A delicate balance act on the tightrope [J]., 2019, 82: 102639.
[3] Giaccone G, van Ark-Otte J, Scagliotti G,. Differential expression of DNA topoisomerases in non-small cell lung cancer and normal lung [J]., 1995, 1264(3): 337-346.
[4] Bax B D, Murshudov G, Maxwell A,. DNA topoisomerase inhibitors: Trapping a DNA-cleaving machine in motion [J]., 2019, 431(18): 3427- 3449.
[5] Jain C K, Majumder H K, Roychoudhury S. Natural compounds as anticancer agents targeting DNA topoisomerases [J]., 2017, 18(1): 75-92.
[6] Kintzios S E. Terrestrial plant-derived anticancer agents and plant species used in anticancer research [J]., 2007, 25(2): 79-113.
[7] Hadavand Mirzaei H, Jassbi A R, Pirhadi S,. Study of the mechanism of action, molecular docking, and dynamics of anticancer terpenoids from[J]., 2020, 40(1): 24-33.
[8] Lee M G, Liu Y C, Lee Y L,. Heteronemin, a marine sesterterpenoid-type metabolite, induces apoptosis in prostate LNcap cells via oxidative and er stress combined with the inhibition of topoisomerase ii and hsp90 [J]., 2018, 16(6): 204.
[9] Lai K H, Liu Y C, Su J H,. Antileukemic scalarane sesterterpenoids and meroditerpenoid from() sp., induce apoptosis via dual inhibitory effects on topoisomerase ii and hsp90 [J]., 2016, 6: 36170.
[10] Otake K, Yamada K, Miura K,. Identification of topoisomerases as molecular targets of cytosporolide c and its analog [J]., 2019, 27(15): 3334-3338.
[11] Skok Z, Zidar N, Kikelj D,. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets [J]., 2020, 63(3): 884-904.
[12] Kaur P, Sharma V. Prospective plant based anticancer lead molecules [J]., 2018, 18(30): 2567- 2583.
[13] Li F, Janussen D, Peifer C,. Targeted isolation of tsitsikammamines from the antarctic deep-sea spongeby molecular networking and anticancer activity [J]., 2018, 16(8): 268.
[14] Kalinski J J, Waterworth S, Siwe Noundou X,. Molecular networking reveals two distinct chemotypes in pyrroloiminoquinone-producingsponges [J]., 2019, 17(1): 60.
[15] Dalvie E D, Gopas J, Golan-Goldhirsh A,. 6,6′- dihydroxythiobinupharidine as a poison of human type II topoisomerases [J]., 2019, 29(15): 1881-1885.
[16] Yang N, Yue R, Ma J,. Nitidine chloride exerts anti-inflammatory action by targeting topoisomerase I and enhancing IL-10 production [J]., 2019, 148: 104368.
[17] Song X D, Wang Y N, Zhang Al,. Advances in research on the interaction between inflammation and cancer [J]., 2020, 48(4):300060519895347.
[18] Almeida A F, Dos Santos C N, Ventura M R. Polyphenols, their metabolites and derivatives as drug leads [J]., 2018, 24(19): 2188-2207.
[19] Jadaun A, Subbarao N, Dixit A. Allosteric inhibition of topoisomerase I by pinostrobin: Molecular docking, spectroscopic and topoisomerase I activity studies [J]., 2017, 167: 299-308.
[20] Xin L T, Liu L, Shao C L,. Discovery of DNA topoisomerase I inhibitors with low-cytotoxicity based on virtual screening from natural products [J]., 2017, 15(7): 217.
[21] Li X, Wu X, Zhu J,. Amexanthomycins A-J, pentangular polyphenols produced byS699ΔrifA [J]., 2017, 102(2): 689-702.
[22] Panter F, Krug D, Baumann S,. Self-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria [J]., 2018, 9(21): 4898-4908.
[23] Hall S R, Toulany J, Bennett L G,. Jadomycins inhibit type II topoisomerases and promote DNA damage and apoptosis in multidrug-resistant triple-negative breast cancer cells [J]., 2017, 363(2): 196-210.
[24] Vann K R, Ekiz G, Zencir S,. Effects of secondary metabolites from the funguson DNA cleavage mediated by human topoisomerase IIα [J]., 2016, 29(3): 415-420.
[25] Alallah M I, Alhemaid F, Bai F,. The binding proximity of methyl β-lilacinobioside isolated fromwith topoisomerase II attributes apoptosis in breast cancer cell line [J]., 2018, 25(8): 1826-1833.
[26] Selvakumar J N, Chandrasekaran S D, Doss G P C,. Inhibition of the atpase domain of human topoisomerase iia on HepG2 cells by 1,2-benzenedicarboxylic acid, mono (2-ethylhexyl) ester: Molecular docking and dynamics simulations [J]., 2019, 19(6): 495-503.
[27] Sośnicki J G, Idzik T J. Pyridones-powerful precursors for the synthesis of alkaloids, their derivatives, and alkaloid-inspired compounds [J]., 2019, 51(18): 3369-3396.
[28] Pommier Y. Topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibitions [J]., 2009, 109(7): 2894-2902.
[29] Mathijssen R H, van Alphen R J, Verweij J,. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) [J]., 2001, 7(8): 2182-2194.
[30] Slatter JG SL, Sams J P,. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following i. V. Infusion of [(14)c] (CPT-11) in cancer patients [J]., 2000, 28(4): 423-433.
[31] Naumczuk B, Wiktorska K, Lubelska K,. New generation of camptothecin derivatives spontaneously alkylating DNA [J]., 2016, 40(9): 7978- 7985.
[32] Jin C, Zhang Q, Lu W. Synthesis and biological evaluation of hypoxia-activated prodrugs of sn-38 [J]., 2017, 132: 135-141.
[33] Wu D, Shi W, Zhao J,. Assessment of the chemotherapeutic potential of a new camptothecin derivative, ZBH-1205 [J]., 2016, 604: 74-85.
[34] Cincinelli R, Musso L, Artali R,. Camptothecin- psammaplin a hybrids as topoisomerase I and hdac dual-action inhibitors [J]., 2018, 143: 2005-2014.
[35] Xu X, Wu Y, Liu W,. Discovery of 7-methyl- 10-hydroxyhomocamptothecins with 1,2,3-triazole moiety as potent topoisomerase I inhibitors [J]., 2016, 88(3): 398-403.
[36] Dai L L, Li D D, Zhao X M,. Synthesis and antitumor effect of sophoridine derivatives bearing an acyclic aryloxy phosphoramidate mustard functionality [J]., 2019, 56(2): 417-425.
[37] Zhao W, Zhang C, Bi C,. Sophoridinol derivative 05d induces tumor cells apoptosis by topoisomerase 1- mediated DNA breakage [J]., 2016, 9: 2805-2817.
[38] Ibric A, Eckerstorfer S, Eder M,. Position-selective synthesis and biological evaluation of four isomeric A-ring amino derivatives of the alkaloid luotonin A [J]., 2019, 24(4): 716.
[39] Rouëssé J, Spielmann M, Turpin F,. Phase II study of elliptinium acetate salvage treatment of advanced breast cancer [J]., 1993, 29A(6): 856-859.
[40] Paoletti C, Le Pecq J B, Dat-Xuong N,. Antitumor activity, pharmacology, and toxicity of ellipticines, ellipticinium, and 9-hydroxy derivatives: Preliminary clinical trials of 2-methyl-9-hydroxy ellipticinium (NSC 264-137) [J]., 1980, 74: 107-123.
[41] Miller C M, O’Sullivan E C, McCarthy F O. Novel 11-substituted ellipticines as potent anticancer agents with divergent activity against cancer cells [J]., 2019, 12(2): 90.
[42] Wang L, Fang K, Cheng J,. Scaffold hopping of natural product evodiamine: Discovery of a novel antitumor scaffold with excellent potency against colon cancer [J]., 2019, 63(2): 696-713.
[43] Zhang X, Rakesh K P, Shantharam C S,. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: A key current imminent needs [J]., 2018, 26(2): 340-355.
[44] Reddy V G, Bonam S R, Reddy T S,. 4beta-amidotriazole linked podophyllotoxin congeners: DNA topoisomerase-IIalpha inhibition and potential anticancer agents for prostate cancer [J]., 2018, 144: 595-611.
[45] Wei J, Chen J, Ju P,. Synthesis and biological evaluation of 4β--acetylamino substituted podophyllotoxin derivatives as novel anticancer agents [J]., 2019, 7: 253.
[46] Woodard J L, Huntsman A C, Patel P A,. Synthesis and antiproliferative activity of derivatives of the phyllanthusmin class of arylnaphthalene lignan lactones [J]., 2018, 26(9): 2354-2364.
[47] Ballinari D, Bonomini L, Ermoli A,. Aurones as telomerase inhibitors: Italia, EP2002/004191 [P]. 2002-04-15.
[48] Priyadarshani G, Nayak A, Amrutkar S M,. Scaffold-hopping of aurones: 2-arylideneimidazo [1,2-a] pyridinones as topoisomerase IIα-inhibiting anticancer agents [J]., 2016, 7(12): 1056-1061.
[49] Priyadarshani G, Amrutkar S, Nayak A,. Scaffold-hopping of bioactive flavonoids: Discovery of arylpyridopyrimidinones as potent anticancer agents that inhibit catalytic role of topoisomerase IIa [J]., 2016, 122: 43-54.
[50] Islam M T. Diterpenes and their derivatives as potential anticancer agents [J]., 2017, 31(5): 691-712.
[51] Chen Y, Zhao J, Li S,. Total synthesis of sesterterpenoids [J]., 2019, 36(2): 263-288.
[52] Esteves-Souza A, Pissinate K, Maciel M,. Synthesis of new-dehydrocrotonin nitrogenated derivatives and their cytotoxic and DNA-topoisomerase I inhibitory activities [J]., 2017, 29(1): 133-139.
[53] Shen B. A new golden age of natural products drug discovery [J]., 2015, 163(6): 1297-1300.
Research progress on antineoplastic topoisomerase inhibitors from natural products
DAI Yi1, 2, SONG Zu-rong1
1. College of Pharmacy, Anhui Xinhua University, Hefei 230088, China 2. Department of Chemistry, University of Science and Technology of China, Hefei 230026, China
Topoisomerase (topo) plays the vital role in solving all of the topological problems of DNA in replication, transcription and other cellular transactions, which is one of the important targets of antitumor drugs. Natural products are an important source of development of topo inhibitors. Exploring topo inhibitors from natural products is one of the hot spots in the development of antitumor drugs. In this paper, the natural topo inhibitors and many derivatives based on natural products are reviewed, in order to provide reference for the development of antineoplastic topo inhibitors.
antitumor; natural products; topoisomerase inhibitors; structure modification; terpene; alkaloids; phenols; quinones
R282.71
A
0253 - 2670(2021)06 - 1785 - 14
10.7501/j.issn.0253-2670.2021.06.029
2020-06-04
安徽省教育厅自然科学研究重点项目(KJ2019A0873)
戴 一(1979—),男,副教授,博士,研究方向为活性天然产物研究。E-mail: daiyiii@163.com
[责任编辑 崔艳丽]
猜你喜欢
新生代(2019年8期)2019-10-28 06:39:30
中成药(2018年2期)2018-05-09 07:20:05
中成药(2017年12期)2018-01-19 02:06:58
中成药(2017年7期)2017-11-22 07:33:25
中成药(2017年8期)2017-11-22 03:18:58
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
合成化学(2015年10期)2016-01-17 08:56:26
中国当代医药(2015年24期)2015-03-01 02:06:12
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
