
PYCR1相关常染色体隐性皮肤松弛症患儿临床表现及遗传学特征分析
2021-03-09 11:49吴文涌上官华坤陈瑞敏
检验医学 2021年2期
吴文涌, 上官华坤, 陈瑞敏
(1.福建医科大学临床医学部,福建 福州 350004;2.福建医科大学附属福州儿童医院内分泌遗传代谢科,福建 福州 350005)
皮肤松弛症又称弹性组织溶解症或早老症,是一种罕见的结缔组织疾病,主要临床表现为早老外观(皮肤松弛、皱纹),根据遗传方式分为常染色体显性皮肤松弛症(autosomal dominant cutis laxa,ADCL)、常染色体隐性皮肤松弛症(autosomal recessive cutis laxa,ARCL)和X连锁皮肤松弛症(X-linked cutis laxa,XLCL)[1]。其中,ARCL发病时间最早,常在婴幼儿期甚至胎儿期出现皮肤受累,除皮肤外观外,还表现出宫内生长迟缓、特殊面容、智力障碍、全面发育迟缓、多脏器结构异常[1-2]。目前,已明确FBLN5、EFEMP2、LTBP4、ATP6V0A2、PYCR1、ATP6V1E1、ATP6V1A1、ALDH18A1为ARCL相关致病基因;根据致病基因的不同,进一步将ARCL分类为Ⅰ型(ⅠA、ⅠB、ⅠC)、Ⅱ型(ⅡA、ⅡB、ⅡC、ⅡD)、Ⅲ型(ⅢA、ⅢB)[3-4]。ARCLⅡB/ⅢB型由PYCR1基因变异导致,被称为PYCR1相关ARCL。PYCR1相关ARCL在我国罕见报道[5]。本研究对1例由PYCR1基因复合杂合变异导致的ARCL及相关文献报道114例PYCR1基因相关ARCL患儿的临床表现、分子遗传学特征进行分析。
1 材料和方法
1.1 研究对象
先证者为男童,2岁2个月,因“生长发育迟缓2年”于2019年8月至福建医科大学附属福州儿童医院内分泌遗传代谢专科门诊就诊。患儿自幼生长较同龄儿迟缓,具体生长速率不详,7个月抬头,1岁3个月独坐,现可扶走,仅能发单音。系第1胎第1产,足月顺产,出生身长不详,体质量1.9 kg[低于同年龄、同性别、同地区新生儿平均体质量3个标准差(<-3s)],就诊前13 d因“双侧腹股沟斜疝、双侧隐睾”于福建医科大学附属福州儿童医院普通外科行“双侧疝囊高位结扎术+双侧睾丸下降固定术”。父母身体健康,否认近亲结婚。
1.2 方法
经患儿父母知情同意后,采集患儿及其父母外周血各2 mL,乙二胺四乙酸抗凝,4 ℃保存待测。采用核酸抽提试剂盒(北京康为世纪生物科技有限公司,批号为CW2087M)抽提所有样本的基因组DNA。采用SureSelect Human All Exon V6试剂盒(美国Aligent 公司)构建基因组DNA的测序文库,采用NovaSeq 6000高通量测序平台(美国Illumina 公司)进行测序。基因检测由中科基因医学检验所完成。采用SIFT软件、Mutation Taster软件、PolyPhen2软件和美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)/美国分子病理学学会(the Association for Molecular Pathology,AMP)指南[6]进行基因致病性分析。采用Sanger测序法对患儿及其父母的变异位点进行验证。
1.3 文献检索
以“PYCR1 AND 常染色体隐性皮肤松弛症”为关键词在万方数据库、维普网及中国知网中检索。以“PYCR1 AND [Autosomal recessive cutis laxa OR ARCL]”为关键词在PubMed数据库检索,检索时间为2009年7月—2020年1月,排除非临床病例报道及临床资料不全的文献。
2 结果
2.1 ARCL患儿的临床资料
患儿,男,2岁2个月,身高81.7 cm[低于同年龄、同性别、同地区儿童平均身高2.48个标准差(-2.48s)],体质量8.0 kg[低于同年龄、同性别、同地区儿童平均体质量2.68个标准差(-2.68s)],上下部量比例正常,反应迟钝,不能理解语言,无法交流与对答,头发稀疏,特殊面容(三角脸,前额隆起、皱纹,鼻翼窄,下颌前突,招风耳),斜颈,胸腹壁静脉显露,躯体皮肤松弛、皱纹明显。心(-),肺(-),腹软。患儿匀称性矮小,排除骨骼发育异常疾病;血常规、尿常规、粪便常规及生化项目、胸片、心脏彩色多普勒超声检查、腹部电子计算机断层扫描未见明显异常,不支持慢性肝脏疾病或慢性肾脏疾病。建议行甲状腺、胰岛素样生长因子1等激素检测,家属拒绝。鉴于患儿身材矮小,合并早老外观、特殊面容及全面发育迟缓,考虑早老症,予基因检测。
2.2 ARCL患儿基因检测结果及其致病性分析
患儿PYCR1基因(NM_006907)存在2个杂合变异,包含1个移码变异c.345delC(p.Arg115Glyfs*7)和1个错义变异 c.413G>A(p.Gly138Asp)。Sanger测序验证显示c.345delC来自父亲,c.413G>A来自母亲,构成复合杂合,见图1。其中p.Arg115Glyfs*7人群频率为0.0004(rs758601634),为已报道过的致病变异(PVS1+PS3+PM2+PP3+PP4)[7-8];p.Gly138Asp变异为未报道的新变异,人群频率为0,SIFT、Mutation Taster和PolyPhen2软件预测为“有害性变异”,根据ACMG/AMP指南,该变异位点为“可能致病性变异(PM1+PM2+PM3+PP3+PP4)”。
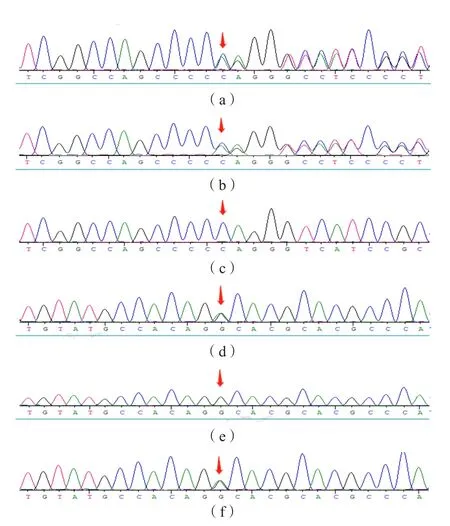
图1 PYCR1基因Sanger测序图
2.3 文献复习
共检索到16篇文献,其中中文1篇、英文15篇,结合本研究的1例患儿,共计115例PYCR1相关ARCL患者,其主要临床表现见图2。
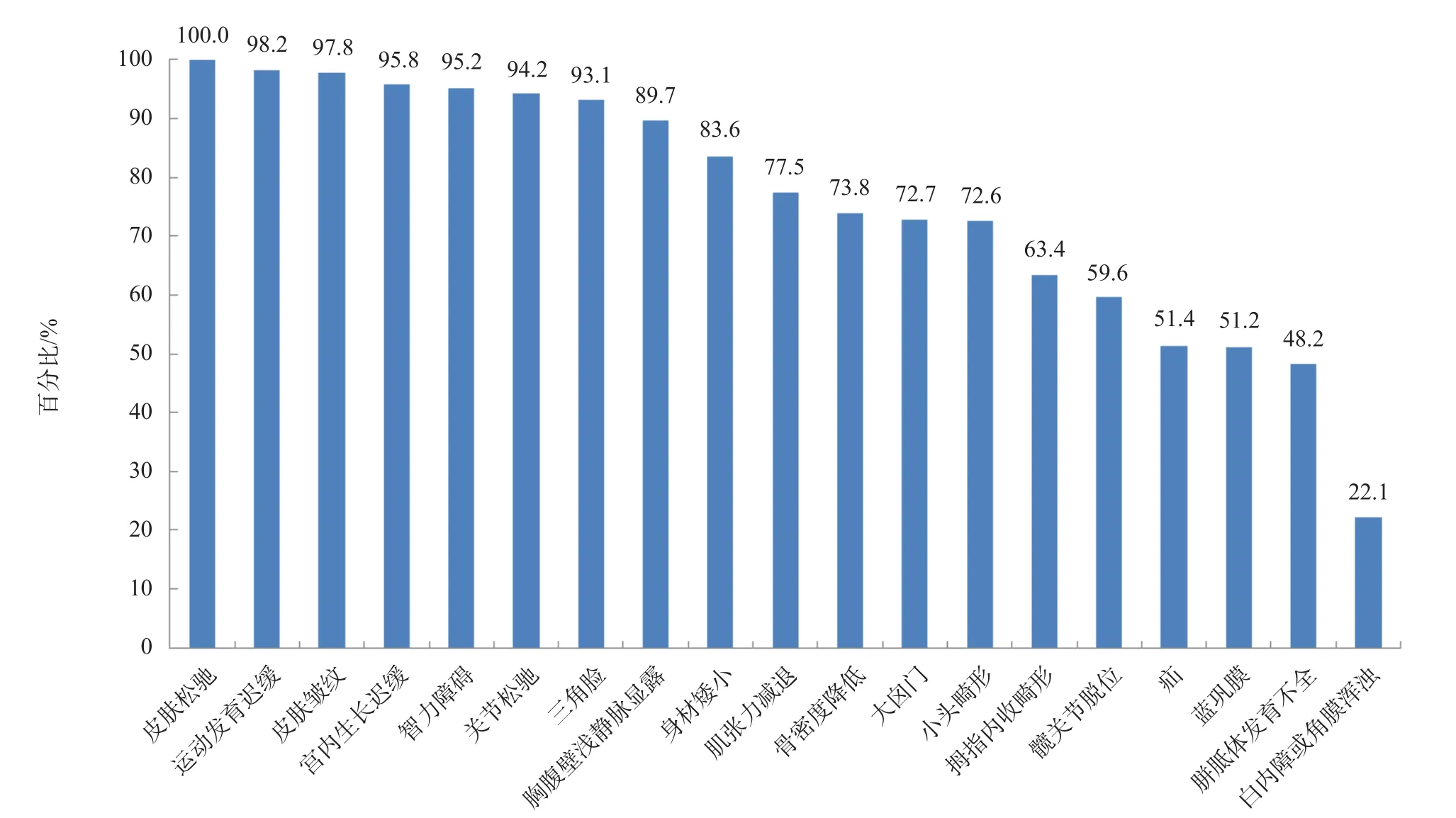
图2 115例PYCR1基因相关ARCL患者的临床表现汇总
115例患者中有98例为纯合变异,有17例为复合杂合变异;共包括42个不同变异位点,其中错义变异27个、剪接位点变异6个、移码变异6个、无义变异2个、内含子变异1个。突变类型与临床表现无明显相关性。
3 讨论
PYCR1基因相关ARCL由GUERNSEY等[9]于2009年首次报道,主要临床表现为早老外观、特殊面容(三角脸、小头畸形)、智力障碍、全面发育迟缓、关节松弛和身材矮小等。本例患儿出现特殊面容(三角脸,前额隆起、皱纹,鼻翼窄,下颌前突,招风耳)、全面发育迟缓、身材矮小、斜颈、胸腹壁静脉显露、躯体皮肤松弛、皱纹明显、双侧隐睾、腹股沟斜疝,符合ARCL的临床表现。基因检测显示存在p.Arg115Glyfs*7和p.Gly138Asp复合杂合变异,根据ACMG/AMP指南,变异位点的致病性明确,结合其典型临床表现,可明确诊断为PYCR1相关ARCL。
本研究检索到114例PYCR1相关ARCL,加上本研究的1例,共115例,其临床表现主要为:(1)早老外观,皮肤松弛、皱纹明显;(2)智力障碍,全面发育迟缓;(3)骨关节异常,关节松弛,骨密度减少,拇指内收畸形,髋关节脱位;(4)特殊面容,三角脸,小头畸形;(5)身材矮小。因PYCR1相关ARCL的临床表现与一些皮肤和骨骼疾病相似,需要与以下疾病鉴别:(1)Geroderma osteodysplasticum,由GORAB基因变异所致,婴幼儿期发病,皮肤表现限于背、腹及手足,胸部以上受累少见,且智力正常[10];(2)Arterial tortuosity综合征(Arterial tortuosity syndrome,ATS),由SLC2A10基因变异所致,有早老外观表现,但以大动脉病变为特征表现[11];(3)ADCL和XLCL,ADCL由ELN基因变异所致,其皮肤改变常在成年期出现,罕见出现肺动脉狭窄、主动脉瘤、支气管扩张和肺气肿等皮肤外器官受累[12],而ARCL患者发生器官受累时常出现心脏、肺脏结构改变;XLCL由ATP7A基因变异所致,除皮肤表现外,可出现特征性的骨骼异常“枕角”[13]。
ARCL各型都有早老外观(皮肤松弛、皱纹明显)这一共同特征,临床上难以区分,但在器官受累和严重程度上具有一定的异质性,其预后与呼吸、循环等系统受累程度有关。ARCLⅠ型常有多脏器受累,可危及生命,可见颅骨发育异常、囟门闭合延迟、关节松弛、髋关节脱位等骨关节异常和腹股沟疝[2,14]。ARCLⅡ型除皮肤松弛外,可出现特殊面容(前额隆起、弓形眉、眼睑外侧下斜、龋齿),生长发育迟缓和骨关节异常[14-15]。ARCLⅢ型又称为De Barsy综合征,除了早老外观、智力障碍、全面发育迟缓外,还会出现眼部受累,如白内障或角膜浑浊[16]。PYCR1变异患者通常被诊断为ARCLⅡB型,小部分眼部严重受累患者被诊断为ARCLⅢB型。各型ARCL由于临床表现重叠,难以直接鉴别,需要进行基因检测以明确诊断。
PYCR1基因位于17号染色体长臂(17q25.3),编码吡咯啉-5-羧酸还原酶1(delta-1-pyrroline-5-carboxylate reductas,PYCR1)。PYCR1是一种线粒体酶,参与L-脯氨酸生物合成过程,在结缔组织的发育中起重要作用[17]。早老外观是ARCL最重要的表现,所有患者均表现出皮肤松弛或皱纹。有研究结果显示,PYCR1基因活跃表达于人体皮肤及骨关节系统,如发生变异可导致成纤维细胞病变,对氧化应激的耐受能力下降,细胞凋亡率增加,最终引起皮肤表现[18]。除此之外,PYCR1基因变异还可导致骨关节异常、身材矮小、智力障碍、全面发育迟缓等。有研究结果显示,PYCR1基因敲除斑马鱼模型出现脯氨酸合成酶缺乏,无法合成足够的脯氨酸和羟基脯氨酸,结缔组织减少,导致运动功能低下、身长不足、体质量不足和早衰[19]。
目前,ARCL尚无有效治疗或预防疾病进展的方法,因此对症治疗十分重要。对于骨密度降低患者,应用双膦酸盐治疗24个月后可有效纠正患者骨密度,预防自发性骨折继续发生,明显改善患者生活质量[20]。整形手术也可对皮肤外观起到改善作用。长期、定期随访有助于早期发现患者消化、呼吸、循环等系统的并发症,及时治疗,改善患者预后[1]。
综上所述,ARCL表型、分型复杂多样。当患者出现早老外观,合并表现为关节松弛、特殊面容、智力障碍、全面发育迟缓和身材矮小时应考虑PYCR1相关ARCL,基因检测有助于最终确诊,长期随访有助于及时对症治疗,改善患者预后。
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
学苑创造·A版(2022年4期)2022-06-18
阅读(快乐英语高年级)(2022年6期)2022-06-17
小学生优秀作文(低年级)(2022年3期)2022-03-29
家庭影院技术(2021年10期)2021-11-20
风流一代·青春(2021年9期)2021-09-23
小学生优秀作文(低年级)(2020年9期)2020-10-26
幸福·婚姻版(2018年9期)2018-11-26
草原(2018年2期)2018-03-02
疯狂英语·阅读版(2013年2期)2013-03-22
