
PRKAR1A基因突变导致肢端发育不全1型4例分析
2021-03-09 11:49:54覃再隆陈少科沈亦平罗静思
检验医学 2021年2期
谌 飞, 覃再隆, 陈少科, 范 歆, 李 川, 易 赏, 沈亦平, 罗静思
(1.广西壮族自治区妇幼保健院遗传代谢中心实验室,广西 南宁 530002;2.广西壮族自治区妇幼保健院内分泌遗传病专科,广西 南宁 530002)
PRKAR1A基因主要指导蛋白激酶A(protein kinase A,PKA)调节亚单位的合成,是PKA四聚体(2个调节亚单位、2个催化亚单位)的关键组成部分之一。PRKAR1A蛋白具有抑制/激活PKA活化的重要功能,参与调节环磷酸腺苷(cyclic adenosine monophosphate,cAMP)的细胞内物质代谢以及调控细胞的增殖、分化和凋亡[1]。PRKAR1A基因由1个N端调节性二聚化/对接结构域、1个包含抑制位点的连接接头和2个cAMP结合域共同构成。PRKAR1A蛋白本身是一个无活性复合体,与cAMP分子结合后引起可逆的空间构象变化,从而激活PKA并释放出活性催化亚基,是cAMP信号传导的主要介导方式[2]。PRKAR1A基因变异通常导致以黏液瘤、色素沉着和内分泌功能亢进三联征为典型特征的卡尼复合征,临床上常需要与McCune-Albright综合征和库欣综合征鉴别[3]。关于卡尼复合征在心胸外科、皮肤科和肿瘤外科等领域的研究已有大量报道[4-5],但关于PRKAR1A基因变异导致肢端发育不全1型的病例我国罕见报道。肢端发育不全1型是一种以身材矮小、严重短指畸形、面中部发育不良和甲状腺功能异常为主要特征的骨骼发育不良疾病,其他常见的临床表现还包括骨龄提前、肥胖症以及涉及甲状旁腺激素、促甲状腺激素(thyroid stimulating hormone,TSH)、降钙素、生长激素释放激素和促性腺激素的多激素抵抗等[6-7]。本研究拟通过分析PRKAR1A基因突变导致的肢端发育不全1型患儿的实验室指标与疾病表型,旨在为肢端发育不全1型的鉴别诊断和早期干预提供参考。
1 材料和方法
1.1 研究对象
选取2016年3月—2019年12月广西壮族自治区妇幼保健院确诊为肢端发育不全1型的患儿4例,其中男2例、女2例,年龄1~12岁。本研究经广西壮族自治区妇幼保健院伦理委员会批准,患儿监护人均知情同意,并签署知情同意书。
1.2 样本采集及测序
收集4例患儿的临床资料(性别、就诊年龄、出生情况、身高、体质量、头围、面部畸形、牙齿、短指畸形、智力发育)及实验室检测结果[TSH、生长激素(growth hormone,GH)、糖化血红蛋白(glycated hemoglobin A1c,HbA1c)、空腹胰岛素(fasting insulin,FINS)、胰岛素样生长因子1(insulin-like growth factor 1,IGF-1)、胰岛素样生长因子结合蛋白3(insulin-like growth factor-binding protein 3,IGFBP-3)、促卵泡刺激素(follicle stimulating hormone,FSH)、促黄体生成素(luteinizing hormone,LH)、游离三碘甲状腺原氨酸(free triiodothyronine,FT3)、游离甲状腺素(free thyroxine,FT4)、三碘甲状腺原氨酸(triiodothyronine,T3)、甲状腺素(thyroxine,T4)、血铅(plumbum,Pb)、血镉(cadmium,Cd)、总钙(calcium,Ca)、碱性磷酸酶(alkaline phosphatase,ALP)、尿常规]。
采集4例患儿及其父母的静脉全血3 mL,乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)抗凝,采用Lab-Aid 820核酸自动化提取仪(厦门致善公司)及配套试剂提取基因组DNA。采用Qubit 4.0荧光定量仪(美国Thermo Fisher公司)检测提取的DNA浓度。采用SureSelect人类全外显子靶向序列捕获系统V5试剂盒(美国安捷伦公司)构建基因组DNA的测序文库,采用HiSeq2500测序系统(美国Illumina公司)进行高通量测序分析。
1.3 测序数据分析及基因变异位点解读
采用基因组分析工具(the genome analysis toolkit,GATK)[8]对测序数据进行生物信息学分析。采用TGex高通量测序数据分析系统(美国TGex公司)对变异位点进行注释和筛选,依据美国医学遗传学和基因组学学会制定的《遗传变异分类标准与指南》[9]进行变异位点的解读与分析,结合患儿临床表型和致病性预测软件数据,确定变异的致病性等级。对于判断为致病性、可能致病性及临床意义不明的变异位点,使用PRKAR1A基因的NM_002734. 4转录本,采用Sanger测序法对患儿及其家庭成员进行变异位点的二次验证。
2 结果
2.1 4例肢端发育不全1型患儿的临床资料
4名患儿均为足月顺产,无家族史,具有相同的典型临床特征,具体表现为:(1)严重的匀称性身材矮小[低于同年龄、同性别、同地区平均儿童身高3~6个标准差(-3s~-6s)]、低体质量[低于同年龄、同性别、同地区平均儿童体质量2~4个标准差(-2s~-4s)];(2)短肢畸形,手指短而弯曲,指骨、掌骨发育不全,见图1;(3)面中部发育不良,前额突出、眼距宽、长人中、鼻梁塌陷、短球状鼻及小下颌,见图2;(4)甲状腺功能异常,FT3、FT4正常,TSH升高。4例患儿的一般临床资料及实验室检测结果见表1、表2。

表1 4例肢端发育不全1型患儿的一般临床资料

表2 4例肢端发育不全1型患儿实验室检测结果
除以上的共同临床表现之外,不同病例还有各自不同的表现。
病例1:自幼喂养饮食均正常,5岁时发现生长发育明显落后,现青春发育延迟,骨龄落后;左手正位数字化摄影提示左手各掌骨及指骨粗短,干骺端增宽,边缘不规则呈喇叭状,骨骺骨小、不规则,呈锥状被包埋,部分干骺愈合;关节间隙清晰,关节对位关系可,骨龄落后,见图3(a)。
病例2:除面部及手部影像特征[图1(a)、图2(a)]外,垂体磁共振成像(magnetic resonance imaging,MRI)检查提示脱髓鞘病变[图4(a)]。
病例3:除面部及手部影像特征[图1(b)、图2(b)]外,母亲孕期染色体芯片检查未见异常,5个月会抬头,独坐独走时间不详,2岁会说话,提示运动及语言发育迟缓。
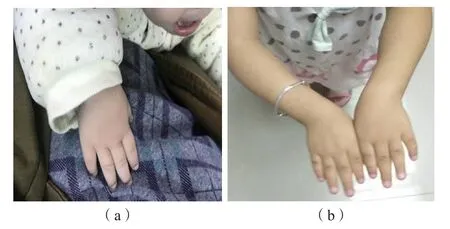
图1 肢端发育不全1型患儿手部特征
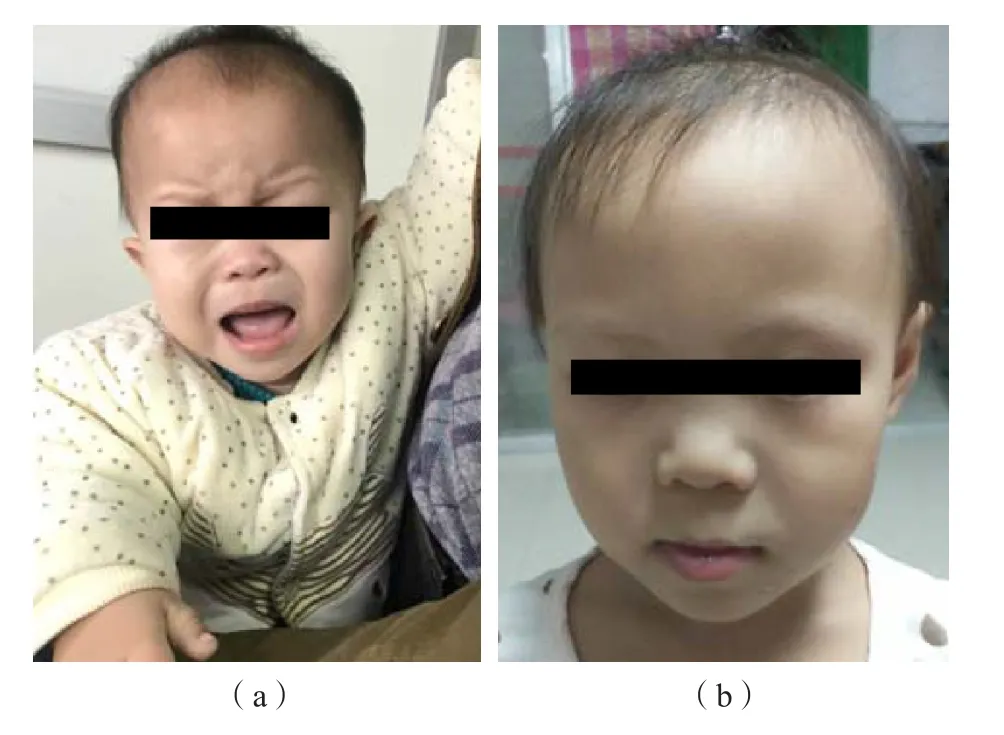
图2 肢端发育不全1型患儿面部特征
病例4:骨龄相符,但左手正位数字化摄影提示明显的肢端骨骼发育异常,见图3(b);垂体MRI未见明显异常,垂体高5.6 mm,脑部双侧卵圆中心多发异常信号,考虑局灶性脱髓鞘病变可能,见图4(b)。
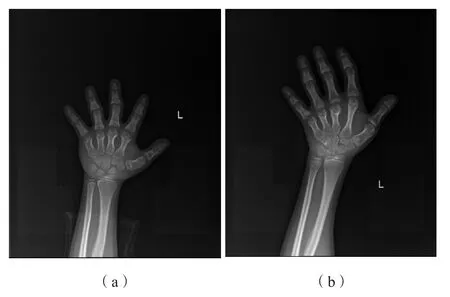
图3 肢端发育不全1型患儿左手正位数字化摄影特征
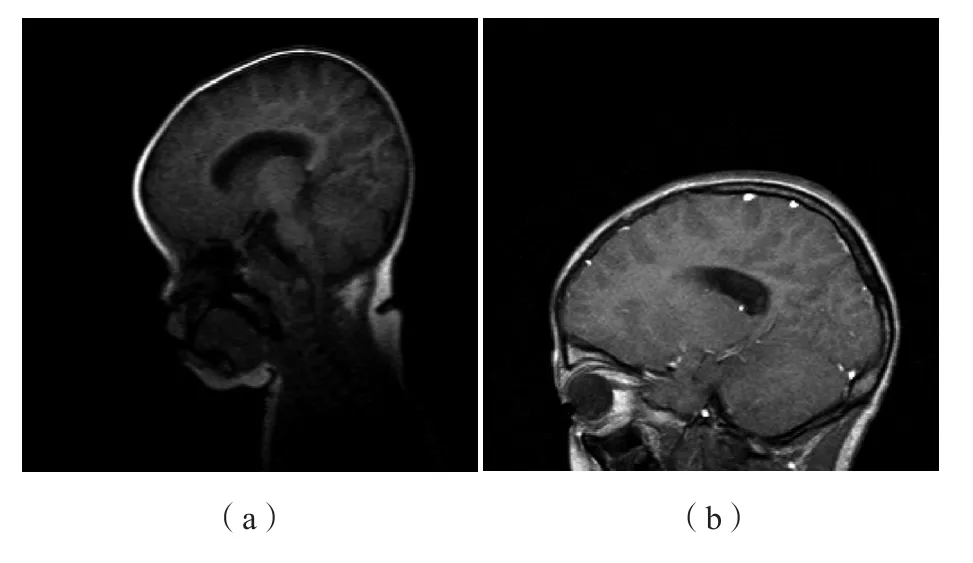
图4 肢端发育不全1型患儿垂体MRI特征
2.2 全外显子高通量测序结果
全外显子高通量测序结果显示,病例1、病例2和病例3均携带PRKAR1A突变(c.1102C>T/p.Arg368*),病例4携带PRKAR1A突变(c.1118A>G/p.Tyr373Cys)。上述突变在人群数据库[the Exome Aggregation Consortium(ExAC)、Genome Aggregation Database(GnomAD)]中均未见报道,在致病数据库(PubMed、ClinVar)中均检索到相似症状的病例报道。经先证者及其父母的Sanger测序二次验证,确认上述突变均为新发突变。根据《遗传变异分类标准与指南》,上述突变均被确定为致病性变异。
2.3 文献已报道的PRKAR1A基因变异种类分析
本研究整理、分析了ClinVar数据库和各类文献报道中明确临床诊断的PRKAR1A变异共计49种,其中引起肢端发育不全1型的12种变异主要分布于结合区域(以cAMP结合区为突变热点区域),包括错义突变11种、无效突变1种;引起卡尼复合征的37种变异则广泛分布于调节区域和结合区域(以调节性二聚体为突变热点区域),包括错义突变8种、无效突变29种,见图5。
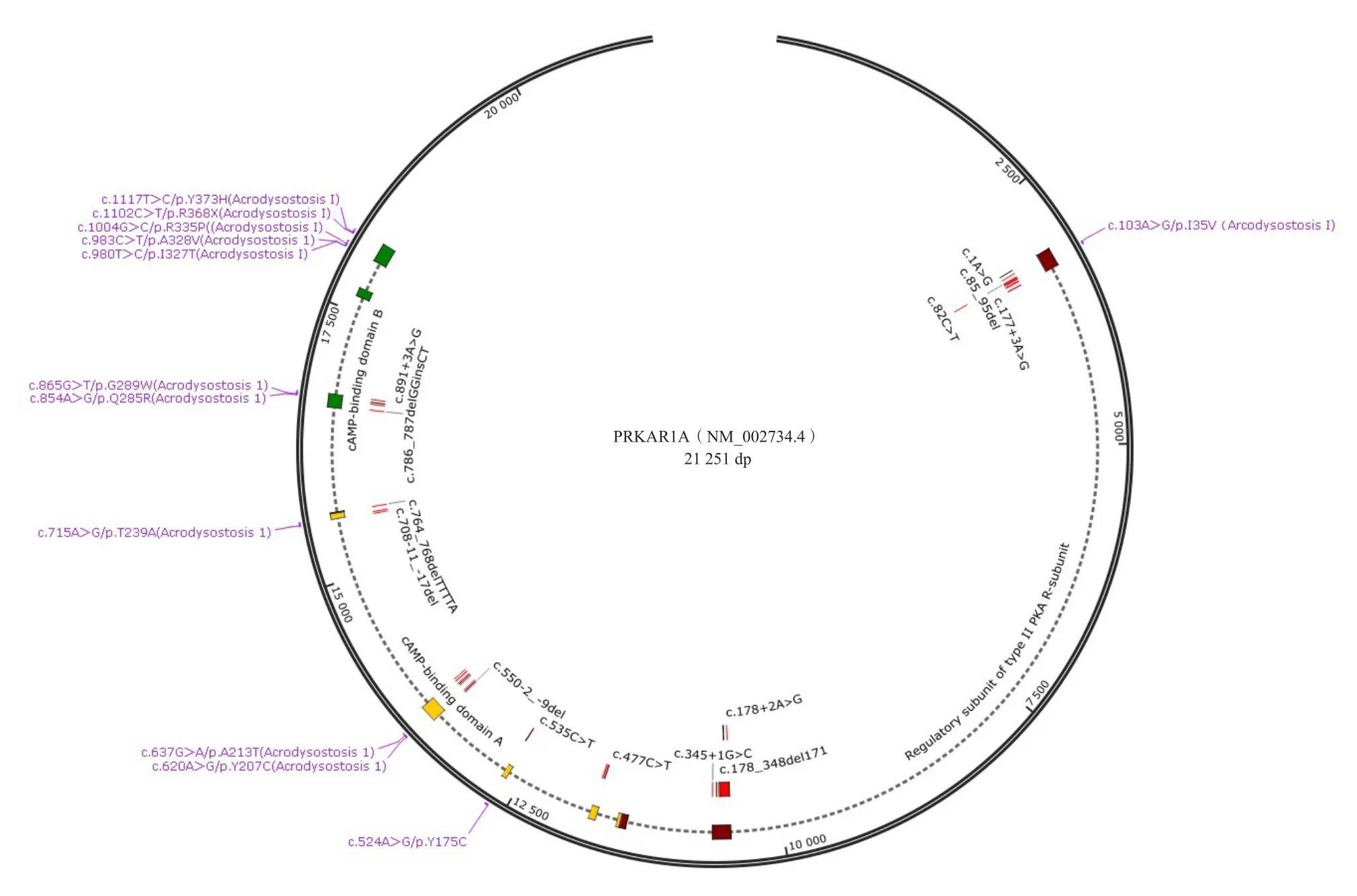
图5 PRKAR1A基因突变基因组位置分布图
3 讨论
本研究统计了2016年3月—2019年12月广西壮族自治区妇幼保健院儿科门诊246例因身材矮小就诊的患儿的全外显子高通量测序数据,检出4例由PRKAR1A变异导致的肢端发育不全1型患儿,临床表现为严重身材矮小、低体质量、短指、特殊面容及不同程度的激素异常等,与目前我国报道的1例肢端发育不全1型患儿的症状类似[10]。国外报道的同一变异位点患儿,多数合并隐睾和肥胖的表现[11-13],但本研究的2例男性患儿并不具有隐睾、肥胖的临床表现,所有患儿均表现为严重的低体质量和生长发育迟缓。以往报道的患儿均有骨龄超前,可能与其肥胖导致早熟相关。但本研究4例患儿中,2例患儿表现为骨龄落后。此外,本研究首次揭示PRKAR1A变异患儿存在生长激素部分缺乏,这对患儿的身高改善及生长激素治疗是否有指导意义有待进一步研究,不同种族及个体间临床症状存在差异的原因也有待深入研究。本研究整理出PRKAR1A突变患儿的实验室指标与疾病表型,旨在完善我国肢端发育不全1型患儿的遗传病诊疗数据,为临床诊断和个体化用药提供理论数据支持。
PRKAR1A基因具备基因多效性,其导致的肢端发育不全1型与卡尼复合征在临床表现上显著不同。PRKAR1A基因同一密码子的不同碱基改变所致的不同氨基酸改变也会导致不同的临床表型。如c.637G>A/p.A213T和c.865G>T/p.G289W会导致以骨骼发育不良为主要表现形式的肢端发育不全1型,而c.638C>A/p.A213D和c.866G>A/p.G289E会导致以多发性黏液瘤和皮肤黏膜色素性病变为主要表现形式的卡尼复合征[12]。卡尼复合征的致病突变机制为功能缺失。ELLI等[13]的研究结果显示,卡尼复合征患儿的PRKAR1A突变体蛋白在表达后会迅速降解,且不与PKA的催化亚基相互作用。PKA原本需要PRKAR1A蛋白结合cAMP才能改变空间构象,从而激活并释放催化亚基。而无义介导的mRNA降解(nonsense-mediated mRNA decay,NMD)使PRKAR1A蛋白失去调控抑制作用,从而在无需cAMP的情况下增加PKA活性,最终导致类似于库欣综合征的临床表现。肢端发育不全1型的致病机制与卡尼复合征完全不同,其突变体蛋白并不会像卡尼复合征患儿的PRKAR1A突变体蛋白一样迅速降解,缺陷的突变体蛋白降低了PKA调节性亚基对cAMP的亲和力,提升了cAMP活化PKA的浓度阈值,最终导致以多激素抵抗和骨骼发育不良为主要症状的肢端发育不全1型。RHAYEM等[12]和LE STUNFF等[14]采用PRKAR1A相同氨基酸的不同错义突变导致的卡尼复合征患儿和肢端发育不全1型患儿构建过表达细胞系,研究基因突变对PRKAR1A蛋白的影响,并在活细胞中利用生物发光共振能量转移检测PRKAR1A突变体蛋白与催化亚基的相互作用,证实cAMP结合缺陷导致PKA激活抗性是肢端发育不全1型常见的分子机制。
综上所述,根据本研究4例PRKAR1A基因突变导致肢端发育不全1型患儿的临床资料,相同基因同一密码子的不同碱基突变可导致不同的临床症状,说明明确患儿的遗传背景才能有针对性地进行对症治疗和预后管理,这也为临床工作中类似病例的诊疗提供了参考。
猜你喜欢
昆明医科大学学报(2021年5期)2021-07-22 07:32:56
天津医科大学学报(2021年2期)2021-03-29 05:30:46
基础医学与临床(2021年9期)2021-03-26 02:11:17
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
百科知识(2015年18期)2015-09-10 07:22:44
中华皮肤科杂志(2014年4期)2014-12-19 12:55:56
雕塑(1996年4期)1996-07-12 07:45:16
