
端羧基水性超支化聚酯促铬吸收及抗氧化的应用
2021-03-09 08:05:32李晨英余佩林陆曼玲刘军陈华林丁克毅
皮革科学与工程 2021年1期
李晨英,余佩林,陆曼玲,刘军,陈华林*,丁克毅
(1.西安工程大学教务处,陕西西安710048;2.西南民族大学化学与环境保护工程学院,四川成都610041)
引言
皮革制品是集美观、舒适、耐用等诸多经济价值于一身的天然优质生物资源,在国内外长期占有庞大而稳定的消费市场[1],铬鞣制革工艺成熟,成本低廉,且成品皮革具有手感丰满、弹性好,机械强度高和稳定性好等不可比拟的优点[2]。因此,铬鞣制革作为目前主流制革工艺,占90%以上的制革市场[3]。我国的皮革制品产量位居世界第一,然而使用最广泛的传统铬鞣工艺中超过30%的铬随废鞣液排污进入环境,使废鞣液中Cr(III)含量高达3000~4000 mg/L。我国每年制革业的废水排放量超过1 亿吨。铬鞣制革的环境污染问题不容忽视[4-6]。因此开发高吸收的铬鞣新材料和新技术一直是清洁化制革的研究热点[7,8]。
除了制革过程带来的污染问题,铬鞣制革另一个值得关注的问题是成品皮革中的Cr(VI)超标。铬鞣制革不可避免的带来皮制品中Cr(VI)的检出[9],而Cr(VI)是国际抗癌研究中心和美国毒理组织公布的致癌物质,并具有水溶性、高毒性和强迁移性。服装箱包等皮革制品经常与人体皮肤接触,其中Cr(VI)就有可能通过污染汗液等方式进入人体,带来健康风险。因此,国内外对皮革中Cr(VI)的含量有严格要求。中国皮革工业协会要求Cr(VI)含量检测作为真皮标志生态皮革四个必检项目之一[10],标准Cr(VI)的残留量要求低于5 mg/kg[11]。目前我国皮制品Cr(VI)超标仍较为普遍,已成为影响我国皮革制品出口的一个主要问题[12]。
皮革中的Cr(VI)主要来源[13]包括:铬鞣剂本身含有少部分Cr(VI)[14]、制革过程使用含铬酸盐染料、皮革使用过程受紫外线、高温、干燥环境因素影响促使Cr(III)氧化,以及最主要的,制革过程使用的加脂剂和油脂中不饱和键氧化形成的过氧化物氧化皮革内Cr(III)形成Cr(VI)[15]。目前,应对皮革中Cr(VI) 形成的最常用方法是使用含酚羟基的植物栲胶,或使用抗氧化剂[16]或还原剂[17]复鞣皮革,但存在影响成品皮革颜色、手感和Cr(VI)去除不彻底等问题[18]。皮革中的Cr(III)被氧化的根本原因之一就是传统铬鞣工艺中Cr(III)在皮胶原中的固定性不好,皮革内存在大量易被氧化的单齿配位和游离的Cr(III)。因此,若能促进皮革内的Cr(III)的吸收和固定,使更多皮革内的Cr(III)能以更为定的配合物形态存在,也就抑制了Cr(III)向Cr(VI)的转化[19]。因此,能促进Cr(III)吸收和固定的高效铬鞣助剂同样也具有抑制Cr3+氧化的效果。
超支化聚合物作为一种新型结构的有机配体,展现出优异的配位能力,与金属离子配位具有容量大、速度快、选择性高等优点[20],加之其合成原料与聚合方法选择范围广,功能基团可调可控,近年来已成为开发清洁化制革新材料的研究热点[21-25]。利用超支化聚合物优异的配位行为,将其作为铬鞣助剂,来增强皮胶原中Cr(III)的吸收和固定,有望在提高铬鞣过程铬吸收率的同时,抑制皮革中Cr(VI)的形成。
本课题组前期已成功合成一系列不同相对分子质量和支化度的端羧基水性超支化聚酯HBP-x(x =1,2…7),并发现其对Cr(III)有良好的配位稳定性[26]。本文将进一步探讨不同相对分子质量和支化度的HBP-x 在水溶液中对Cr(III)氧化的抑制作用,并以HBP-4 为例,作为助剂作用于白皮粉的浸酸铬鞣实验,验证其促铬吸收和固定的作用。
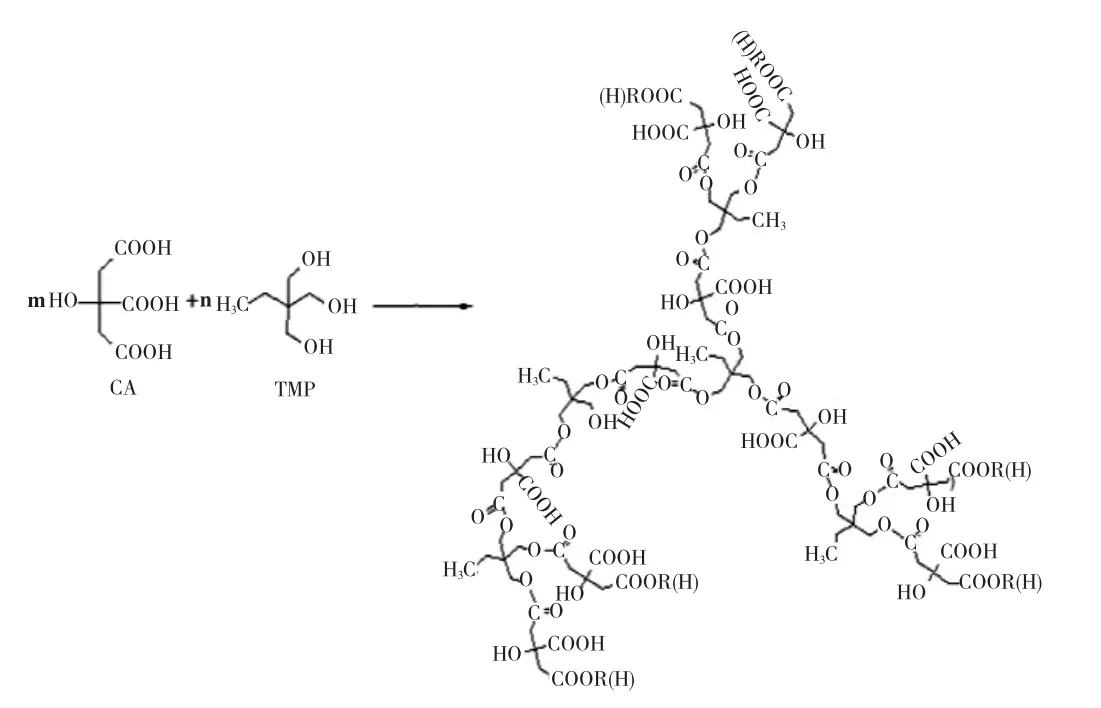
图1 HBP-x(x=1,2…7)的合成过程示意图Fig.1 Route for synthesis of HBP-x(x=1,2…7)
1 试验部分
1.1 试剂与仪器
(1)试验材料
重铬酸钾(AR)、氯化铬(AR)、双氧水(30%)、柠檬酸(AR)和乙酸(AR),甲酸(AR)、硫酸(AR)以上试剂均来自成都市科龙化工试剂厂;聚丙烯酸(BR 级,Mw=2125)Sigma-Aldrich 公司;高吸收铬鞣剂,内蒙古黄河铬盐股份有限公司;白皮粉(AR),中国林业科学研究院林产化学工业研究所科技发展总公司单宁化工实验室,干燥失重12%~14%,pH 值5.0~5.5,吸收单宁能力0.425 g。
水性超支化聚酯HBP-x(x=1,2…7)为实验室自制,以三羟甲基丙烷为核与柠檬酸通过缩合聚合制备[26]。其相对分子质量和支化度等相关参数见表2,其合成过程示意如图1。
(2)仪器
U-2010 型紫外可见分光光度计,HITACHI 公司;差示扫描量热测试仪(DSC Q20,TA),USA,升温速度10 ℃/min,高纯氮氛;电子拉力实验机,济南川佰仪器设备有限公司,型号WNW-5,精度1 级;皮革收缩温度测定仪:中国农业科学院兰州畜牧与兽药研究所;鞣制使用HZS-H 水浴振荡器代替转鼓;Leitz-AMR-1000 型扫描电子显微镜,德国Leitz 公司;PHS-3C 型酸度计,上海磁创益仪器仪表有限公;JB/T10016 型测厚规(量程0~10 mm,精确度0.01 nm):上海恒量量具有限公司。DZF6050 型真空干燥箱:上海精宏实验设备有限公司;热重分析仪TGA Q500 V20.10 Build 36。

表1 白皮粉的浸酸铬鞣与脱铬工艺Tab. 1 Technological process of chrome tanning and dechroming of white hide powder
1.2 双波长吸光光度法测Cr(Ⅵ)浓度工作曲线的绘制
分别配制50 mL 0.1 mol/L CrCl3溶液和25 mL 5.0×10-4mol/L K2Cr2O7溶液。用移液管向编号0~6的10 mL 容量瓶中分别移取5 mL 0.1 mol/L CrCl3溶液 和 0、1.0、2.0、3.0、4.0、5.0 mL 5.0 ×10-4mol/L K2Cr2O7溶液,用蒸馏水定容摇匀,25 ℃下测定溶液在λ1=378 nm,λ2=625 nm 两个波长下吸光度的差值ΔA,并以ΔA 为纵坐标,Cr(VI)的摩尔浓度为横坐标绘制双波长吸光光度法测Cr(VI)浓度的工作曲线。
1.3 超支化聚合物参与下Cr(III)的氧化实验
取50 mL 烧杯编号后向其中移取12.5 mL 0.2 mol/L CrCl3溶液,实验组为1~7 组,分别向其中加入HBP-1、HBP-2、HBP-3、HBP-4、HBP-5、HBP-6、HBP-7 七种配体各0.067 g(占CrCl3质量的10%),对照组为8~10 组,第8 组添加等质量线性聚丙烯酸(PAA,Mw=2125),第九组添加等质量柠檬酸(CA),第10 组为空白对照。各组用酸度计调节溶液pH=4.0。分别添加将上述溶液移入25 mL 容量瓶中,并用蒸馏水定容,室温下静置12 h,使配体和Cr(III) 离子进行配位反应。用移液管从容量瓶中取5 mL 溶液于15 mL 离心管中,向其中加入5 mL pH=2.0 的15%H2O2,摇匀后于室温下静置6 h,使其中Cr(III)氧化。6 h 后,将离心管置于50 ℃烘箱中静置3 h 以分解体系中未参与反应的H2O2。随后使溶液降温至25 ℃,采用双波长分光光度法测定式样在378 nm 和625 nm 两个波长下吸光度的差值ΔA。并通过上文中的工作曲线计算各组中氧化产物Cr(VI)的浓度。
1.4 HBP-4 用于白皮粉浸酸铬鞣的效果
实验组取2 g 白皮粉于150 mL 锥形瓶中,移液管移取pH=2.8、25 mL 1 mol/L NaCl 水溶液,25 ℃于水浴振荡器上以150 r/min 摇动,并缓慢多次滴加10%的甲酸水溶液,添加总量0.4 g,再缓慢多次加入质量分数5%的硫酸,直至pH 稳定在2.8~3.0 以此作为浸酸操作。向实验组中添加0.5 g 市售铬鞣剂和0.075 g(占铬鞣剂质量15%)的HBP-4,对照组不添加HBP-4,两组均按照表2 的工艺模拟铬鞣。将实验组和对照组鞣后皮粉40 ℃真空干燥。将鞣后皮粉块取样做示差扫描量热分析来测定皮粉的变性温度;通过热重分析测其分解温度;通过干燥皮粉块新鲜断面喷金处理做扫面电镜来观察两组内部胶原纤维形貌;通过对鞣后皮粉块用甲酸进行脱鞣,来考察实验组和对照组Cr(III)在皮胶原中的固定情况。本实验中白皮粉的浸酸铬鞣工艺和皮粉的脱鞣工艺见表1。
2 结果与讨论
2.1 HBP-x 与Cr(III)配合物的抗氧化稳定性
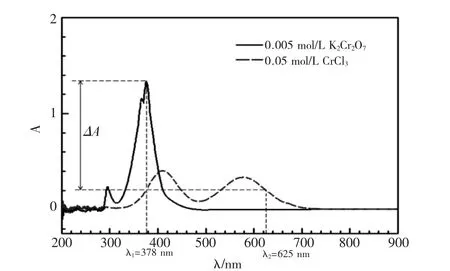
图2 0.1 mol/L CrCl3 和0.02 mol/L K2Cr2O7 的紫外-可见光谱Fig. 2 UV-vis absorbance spectra of 0.1 mol/L CrCl3 and 0.02 mol/LK2Cr2O7 aqueous solution
本文通过双波长分光光度法来检测HBP-x(x=1,2…7)作用下Cr(Ⅲ)体系经H2O2氧化后体系中Cr(VI)浓度。利用水溶液中Cr(III)和Cr(VI)的浓度和吸光度分别在0~5.0×10-2mol/L 和0~2.5×10-4mol/L 具有很好的线性[28],双波长分光光度法可以简便高效检测体系中Cr(VI)含量[29],并有不亚于国家标准方法[30]“二苯碳酰二肼分光光度法”的准确度[31]。

表2 各组经H2O2 氧化后的Cr(VI)浓度Tab. 2 Cr(VI) concentrations in each group after H2O2 oxidation
图2 为本实验测得Cr(III)和Cr(VI)混合水溶液的紫外-可见光谱,CrCl3在λ1和λ2处的吸光度相同,而K2Cr2O7在λ1处吸光度很大、λ2处吸光度为0,当单位时间内分别用波长为λ1和λ2的两束单色光照射混合液,其λ1和λ2处吸光度的差值ΔA 就和Cr(VI)的浓度成正比。只需做相应的工作曲线就能通过ΔA 求出体系中Cr(VI)的浓度。
图3 为通过双波长分光光度法绘制的测定Cr(VI)的工作曲线,线性良好。下文中,通过式2 即可计算氧化实验中各组氧化后体系中Cr(VI)的含量。

氧化试验中各组经H2O2氧化后由双波长分光光度法测得的体系中Cr(VI)的浓度见表2,体系中Cr(VI)的浓度越低说明其体系抗氧化能力越强。具体所的结果分析如下:
(1)添加端羧基超支化聚酯HBP-x(x=1,2…7)可有效抑制体系中Cr(III)离子的氧化。实验组1~7 组中Cr(III)被氧化的程度整体偏低,Cr(VI)浓度最低只有1.441×10-5mol/L,远低于添加线性聚丙烯酸和小分子配体柠檬酸的对照组,而不添加羧酸根配体的空白组第10 组,体系中Cr(VI)浓度高达7.553×10-5mol/L,数倍于实验组。说明在Cr(III)水溶液中引入羧酸根配位可以有效的稳定Cr(III)离子,抑制其氧化。端羧基超支化聚酯HBP-x 稳定Cr(III)抑制氧化的能力明显优于同相对分子质量同含大量羧基的线性聚丙烯酸。而柠檬酸分子结构中带有三个羧基,水溶性好,配位能力强,和金属离子络合过程可生成稳定的环状螯合结构,配合物稳定性好,是铬鞣剂中常用的蒙囿剂原料。表2 中可见柠檬酸做配体的第9 组,其稳定Cr(III)抑制氧化能力优于含大量羧酸根的PAA,但比起除第2 组以外的其余实验组则明显较差。说明具有超支化结构的端羧基聚酯HBP-x(x=1,2…7)得益于其分子结构优势,对Cr(III)有更强的配位能力和抗氧化能力。

图3 双波长分光光度法测Cr6+浓度的工作曲线Fig.3 Working curve of Cr(VI) concentration measured by dual wavelength spectrophotometry
(2)HBP-x 支化度越大,其稳定Cr(III)抑制其氧化的能力就越好。HBP-1、HBP-5、HBP-6、HBP-4 的质均相对分子质量都在1000-2000 之间,支化度从0.43 至0.72 增大。添加这四种配体的1、5、6、4 实验组,体系中Cr(VI)浓度随配体支化度的增加而减小。这主要因为支化度大的配体更容易在和中心离子Cr(III)配位过程中形成桥键和环状结构,有效提高了Cr(III)的稳定性。
(3)HBP-x 相对分子质量较高时,其稳定Cr(III)抑制其氧化的能力大幅降低。HBP-7、HBP-3、HBP-4、HBP-2 的支化度都在0.7 左右,但相对分子质量依次增大。添加这四种配体的第7、3、4、2 组,体系Cr(VI)浓度在低相对分子质量的3、4、7 组均远低于8、9 两对照组,而添加配体相对分子质量最高的第2 组,产物Cr(VI)浓度最高,接近8、9 两对照组。可认为配体相对分子质量对配合物抗氧化能力的影响交大,原因主要是,大相对分子质量大体积的配体形成的配合物稳定性差,易解离,因此溶液中游离Cr(III)浓度较其他实验组更大,Cr(III)也就更容易被氧化。且Cr(III)的氧化反过来更促进了配合物解离平衡的移动。因此,制备用作铬鞣添加剂的超支化聚合物,必须严格控制产物保持在低聚状态,相对分子质量以2000 左右为宜。

表3 白皮粉的浸酸铬鞣实验及脱鞣实验结果Tab. 3 Experiment results of pickling and chrome tanning of white hide powder
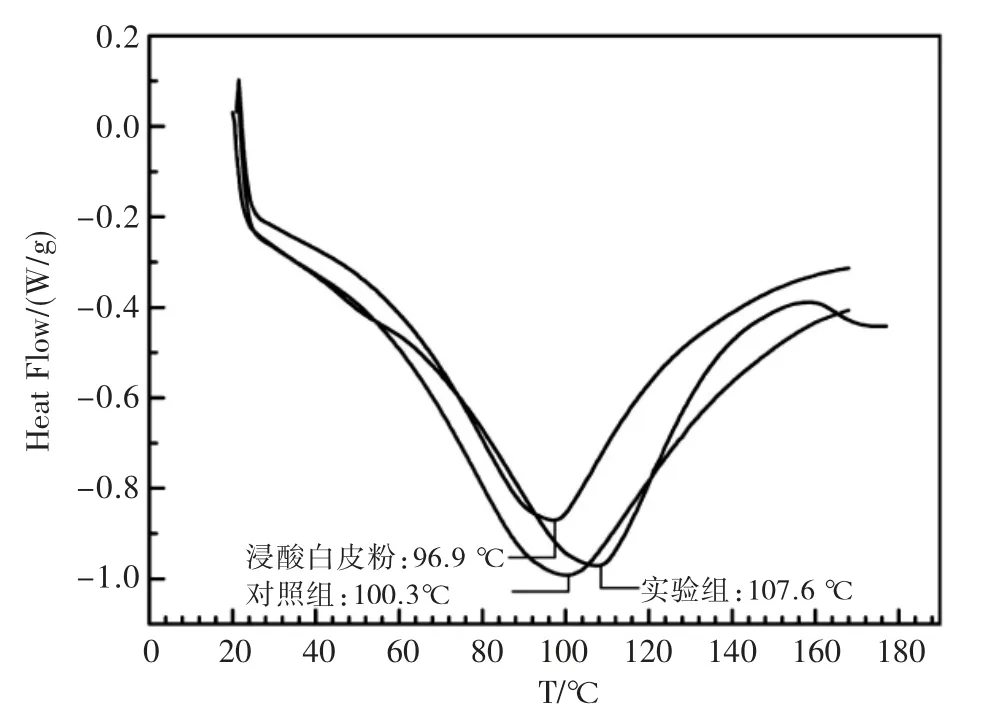
图4 浸酸白皮粉、实验组和对照组鞣后皮粉的DSC 图谱Fig.4 DSC spectrum of white hide powder in experimental group and control group
表2 中第4 组使用支化度较高、相对分子质量适宜的HBP-4,其体系中Cr (VI) 浓度最低,只有1.441×10-5mol/L,远低于添加柠檬酸和线性聚丙烯酸的对照组8,9,证明端羧基的超支化聚酯HBP 的配位能力更优于线性聚羧酸和小分子多元酸,并由显著的抑制Cr(III)氧化的能力。因此,下文中以HBP-4 为例,通过白皮粉模拟浸酸铬鞣来考查端羧基超支化聚合物用于铬鞣过程,促进铬在皮胶原纤维中吸收和固定的能力。
2.2 添加超支化聚合物的白皮粉浸酸铬鞣实验
表3 中的数据充分说明HBP-4 的使用促进了Cr(III)在皮胶原纤维中的吸收和固定。白皮粉由灰皮经机械作用打碎成粉后脱水制得,不像酸皮有紧致的表面和一定的厚度。皮粉具有丰富而裸露胶原纤维和极大的比表面积,可以使胶原纤维和铬鞣浴液充分接触,不存在渗透过程,其铬吸收率远高于实际的酸皮铬鞣情况。使用白皮粉做浸酸铬鞣,可较好地考查Cr(III)在胶原纤维见的固定情况。由HBP-4 参与的实验组白皮粉铬吸收率达86.2%,高于对照组的82.8%。更为重要的是,两组鞣后皮粉在甲酸脱鞣和离心操作这样强力的化学和物理脱鞣作用下,实验组上清液在427 nm 处吸光度为0.006,而未添加HBP-4 的对照组,吸光度高达0.064,反映出二者上清液中Cr(III)浓度存在数量级差异,充分证明使用HBP-4 的实验组,Cr(III)在皮胶原中得到更好地固定。
究其原因,当含有丰富末端官能团的超支化聚合物加入到铬鞣浴液中时,配位能力强的末端基团可以与Cr(III)快速络合,使大量铬核被携带于超支化聚合物的分子链的末端,以一个含多个铬核的大分子配合物的形式整体进入皮胶原内。这样,即使超支化聚合物上络合的铬核只有少部分能和胶原纤维结合,也能保证整个配合物大分子稳定的固定在皮内;同时,超支化聚合物本身大量的端基官能团也可以与胶原纤维相互作用,通过氢键等形式促进配合物整体在皮内的固定。多方面促进Cr(III)在皮内的结合强度,减少铬鞣废水中铬含量,达到提高成革品质和环保的双重效果。

图5 实验组(a)和对照组(b)皮粉的TG 曲线Fig.5 TG spectrum of white hide powder of experimental group (a) and control group (b)
图4 为表3 中两组鞣后皮粉和未经铬鞣的浸酸白皮粉真空干燥后取样通过DSC 测得的热变性温度。未经铬鞣的“浸酸白皮粉”样品热变性温度为96.9 ℃,经铬鞣后的对照组,皮粉的热变性温度提升至100.3 ℃,而在铬鞣过程中添加聚合物HBP-4 的实验组具有更好的热稳定性,其热变性温度达到107.6 ℃,明显优于对照组。图5 为皮粉的热重实验,铬鞣过程添加HBP-4 的实验组(a)皮粉,其分解温度也比未添加HBP-4 的对照组(b)高出4 ℃,更加印证了HBP-4 的添加可以提高鞣后皮粉的热稳定性。

图6 实验组(a)和对照组(b)皮粉的SEM 照片Fig.6 SEM photos of hide powder of experimental group (a) and control group (b)
图6 为表4 中实验组和对照组经真空干燥的鞣后皮粉块新鲜断面处放大40 倍的SEM 照片,观察其中胶原纤维的存在形态可明显看出,添加HBP-4 的实验组(a)胶原纤维间的交联更为紧密,相互聚集成束,直径较粗。而对照组(b)纤维束松散且直径较细。从中可以直观的发现,超支化聚合物的添加可以提高铬鞣铬中胶原纤维的之间的交联密度。也解释了添加HBP-4 的实验组皮粉鞣后更优的热稳定性。
3 结论
端羧基水性超支化聚酯HBP-x 在Cr(III)水溶液中的使用明显提高了体系的抗氧化稳定性的影响。考查HBP-x 分子结构对其抑制Cr(III)氧化的影响发现,其抗Cr(III)氧化能力随HBP-x 支化度的增高而增高;但在相对分子质量较高时,抗氧化能力有明显降低。而分子具有较高的支化度,且质均相对分子质量在2000 左右的端羧基超支化聚酯HBP-4 对抑制Cr(III)氧化具有最佳效果。同时,白皮粉浸酸铬鞣实验证明,使用HBP-4 作为助剂可有效提高鞣制过程胶原纤维的交联密度,促进Cr(III)在胶原纤维中的吸收和固定。
猜你喜欢
小资CHIC!ELEGANCE(2021年32期)2021-09-18 06:17:14
发明与创新·小学生(2018年2期)2018-02-07 10:49:06
小学阅读指南·低年级版(2017年5期)2017-05-18 11:21:04
合成化学(2015年3期)2016-01-17 09:02:05
Coco薇(2015年12期)2015-12-10 02:44:33
应用化工(2014年10期)2014-08-16 13:11:29
应用化工(2014年7期)2014-08-09 09:20:23
文苑·感悟(2014年3期)2014-03-28 08:12:02
华东理工大学学报(自然科学版)(2014年6期)2014-02-27 13:49:40
河北医科大学学报(2011年11期)2011-03-25 10:17:21
