
单原子催化剂Ir1 /MoS2表面上NH3吸附理论研究
2021-03-08 11:15:06肖香珍银召利张建伟
河南科技学院学报(自然科学版) 2021年1期
肖香珍,银召利,张建伟
(1.河南科技学院实验管理中心,河南新乡453003;2.河南科技学院新科学院,河南新乡453003)
贵金属虽然具有良好的催化活性,但因贵金属催化剂的利用效率低[1-7],往往增加了催化成本.最近的研究发现单原子催化剂可以实现原子利用率的最大化,并有着高选择性、高活性和高稳定性的明显优势[8-14],被认为是一种潜在的替代现有贵金属催化剂的材料.通常单原子催化剂需要选择合适的载体,单原子可以吸附在载体表面或嵌入到载体结构中形成催化剂.目前选用的载体大多是金属及其金属氧化物,最近研究显示二维材料MoS2、石墨烯[15-16]、六方氮化硼(h-BN)[17]等作为载体有很大的应用前景.之前本团队曾采用第一性原理方法,多次研究小分子在过渡金属Ir催化剂表面的吸附性能,且得出了较好的结果[18-21].因此,考虑到贵金属Ir对NH3的解离有很好的低温催化活性及较好的产物选择性,选择以单层MoS2作为载体,以单原子Ir吸附在二维材料MoS2表面上来形成单原子催化剂Ir1/MoS2,而目前关于NH3在单原子催化剂Ir1/MoS2上吸附的理论研究报道较少[22-23].因此,通过理论计算模拟出近似真实的催化剂表面,研究了NH3在单原子催化剂Ir1/MoS2表面的吸附行为,为寻找出活性高的催化剂,又能减少贵金属的使用量,实现贵金属原子利用率的最大化提供一定的理论依据.
1 计算模型和方法
MoS2是一种具有二维层状结构的新型半导体材料[24],晶体结构类似于石墨烯,具有二维层状结构,每一层都是三明治式SMoS的结构,三个原子层中的原子都按平面六角阵列方式排列,其作为载体应用前景较大.
本工作中单原子催化剂Ir1/MoS2表面采用Slab超胞模型模拟,所建模型选取3×3周期性单层MoS2超胞.Slab模型如图1所示,图1-A、图1-B分别是周期表面模型的俯视图和侧视图.原子间距结构优化后本征单层MoS2的晶胞参数为0.319 nm,与实验值0.316 nm[25]接近.
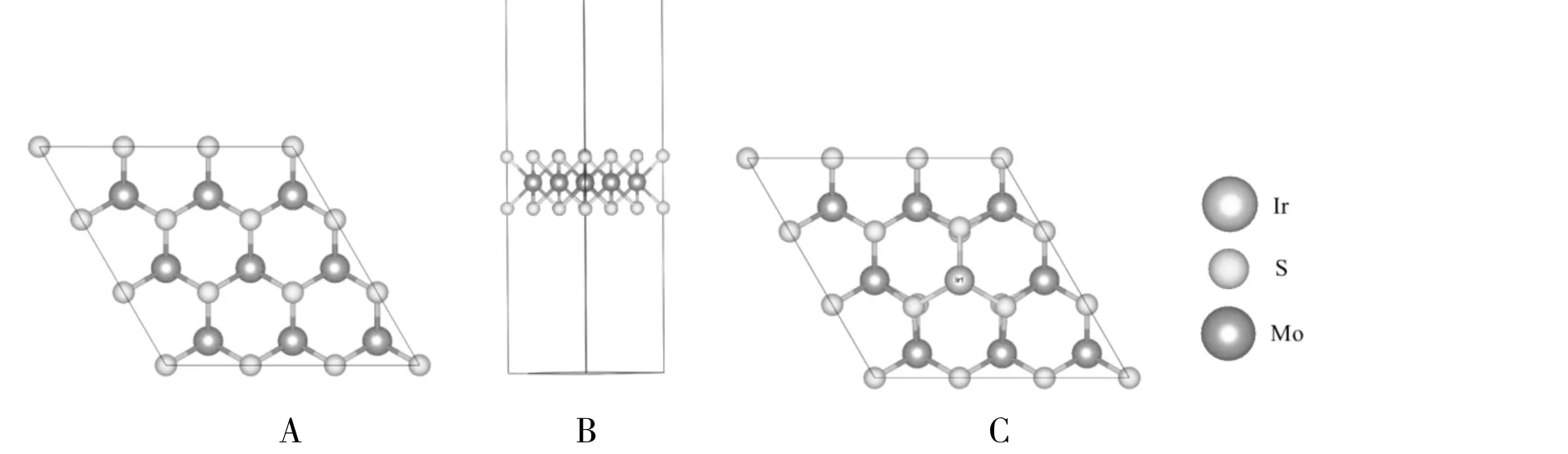
图1 单层MoS2超胞的俯视图和侧视图及单原子催化剂Ir1/MoS2模型Fig.1 Top view and side view of single layer MoS2supercell and the model of single-atom catalysts Ir1/MoS2
采用基于密度泛函理论(DFT)[26-27]方法的VASP[28-29]软件包,离子芯势采用PAW方法描述[30],采用平面波基组展开,交换关联泛函采用GGA-PW91[31]形式,截断能为500 eV,不可约布里渊区积分采用Monkhorst Pack方案[32],K点网格密度取为5×5×1.在计算过程中考虑表面弛豫,固定下面2层原子,吸附物NH3和Ir放在表面S一侧,真空层厚度取为1.5 nm.通过频率分析对所有优化得到的最优构型加以验证,即局域极小点对应全部实频.
2 结果与分析
2.1 过渡金属吸附Ir在单层MoS2载体上的落位
单原子可以附着在载体表面或嵌入载体形成单原子催化剂,根据MoS2的结构考虑了Ir原子分别附着在四重空位(fcc位)和三重空位(hcp位)两个典型的吸附位点.通过计算和比较不同吸附构型的吸附能来确定体系Ir1/MoS2的稳定性结构.将MoS2视为吸附表面,Ir原子为吸附物,吸附能通过下面方程计算得到单个Ir原子附着在MoS2表面的四重空位(fcc位)的吸附能为Eads=2.98 eV,hcp位为Eads=3.98 eV,频率分析得到hcp位对应全部实频,无虚频,显示hcp位相对稳定,其结构俯视图示于图1-C.

2.2 NH3在体系Ir1/MoS2上吸附
2.2.1 几何结构 NH3分子含有孤对电子,在表面上应为给电子吸附.对于NH3在体系Ir1/MoS2上的吸附,考察了其在单原子Ir上的不同吸附构型,如图2所示,图中每一格左边为吸附后的俯视图,右图为侧视图.此时吸附能定义为
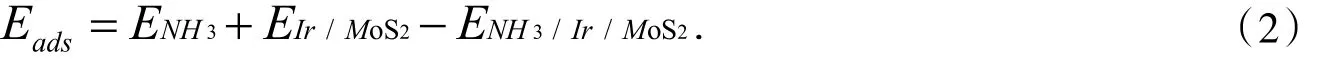
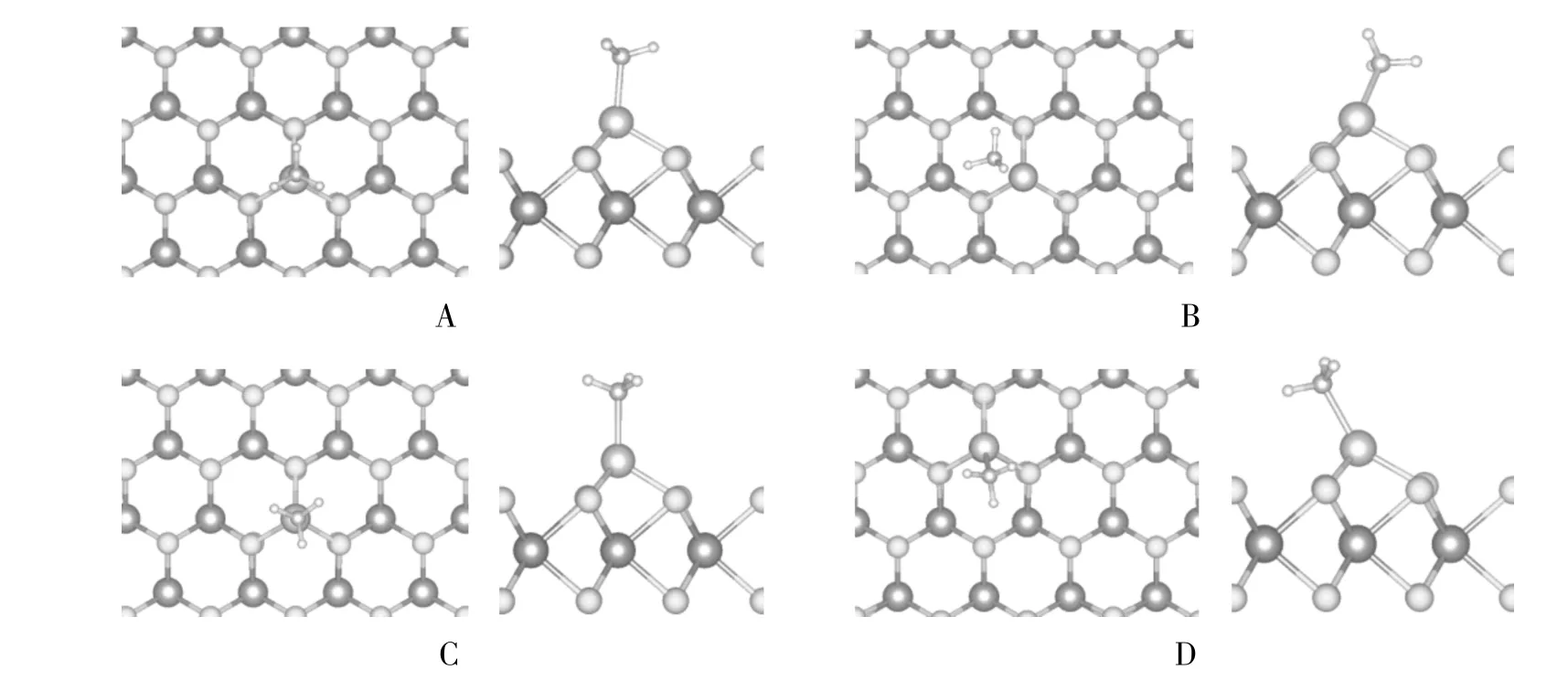
图2 NH3在体系Ir1/MoS2上吸附的构型Fig.2 The configuration diagram of NH3adsorption on Ir1/MoS2system
图2-A中NH3分子通过N原子和MoS2表面上的Ir原子相连,它的C3垂直于体系Ir1/MoS2,三个N-H键指向远离表面的方向,且投影至Ir-S键方向.图2-C也为垂直结构,但三个N-H键是投影至六角表面的fcc位,图2-A和图2-C两种构型吸附能均为1.58 eV,计算结果如表1所示.Ir-N和N-H键长(dIr-N、dN-H)以及夹角∠HNH 也几乎相同,与气相 NH3构型(dN-H=0.102 nm,∠HNH=107.3°)比较无明显变化,可见吸附之后NH3分子结构没有发生明显改变,说明内层轨道在吸附过程中受的扰动非常小,基本保持了其在气态分子中的特征.图2-B和图2-D均为倾斜结构.计算显示图2-B结构中倾斜方向上N、Ir原子与最近的S原子之间的两个夹角∠NIrS分别为 100.4°、97.9°,图 2-D 结构中倾斜方向上两个∠NIrS分别为 108°、100.7°.通过计算吸附能,图2-B、图2-D结构的吸附能分别为1.61 eV、1.63eV,说明图2-D的结构更稳定.这个最稳定吸附位结构类似于NH3在Ir(110)上吸附结构[19],而NH3在贵金属Ir(110)面上的吸附能为1.11 eV,实验研究所用催化剂为单原子催化剂,相比较贵金属Ir作为催化剂,减少了贵金属的使用量,大大节省了资源.
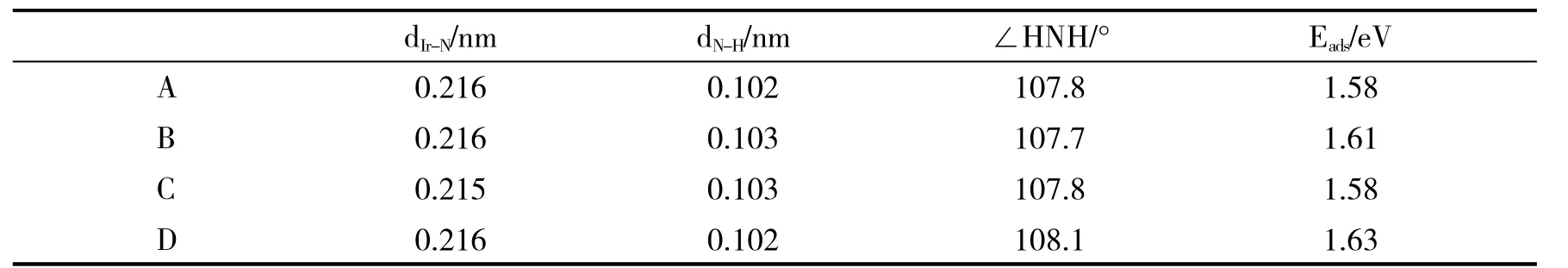
表1 NH3在Ir1/MoS2体系上吸附不同构型的几何参数Tab.1 The geometrical parameters of NH3adsorbed on Ir1/MoS2system
2.2.2 NH3在Ir1/MoS2体系上最稳定吸附位的电子结构分析 为弄清楚NH3吸附前后电荷转移情况,体系的差分电荷密度计算如下
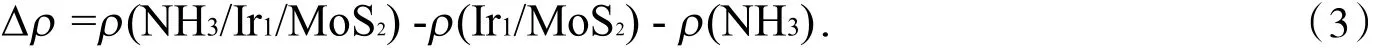
通过分析电荷密度差分图,发现N原子的上下方以及Ir原子上下方出现电荷的降低,如图3-C,这说明在Z方向上NH3和吸附位点金属Ir的部分轨道的电荷重新分布到了N原子及Ir的其他轨道上,而气相NH3分子的3a1轨道是由N原子的2Pz轨道形成的,易在Z方向上发生电荷迁移.图3-B中N原子与吸附点Ir原子中间部分电荷出现显著增加,如图3-A箭头所示,说明电荷转移到了N原子与吸附点Ir原子的中间,这正是NH3稳定吸附于体系Ir1/MoS2的主要原因.另外,Ir原子的x、y方向上的各轨道与NH3各轨道间无明显相互作用,但是有少量电荷积聚,这是由其他态迁移过来的.
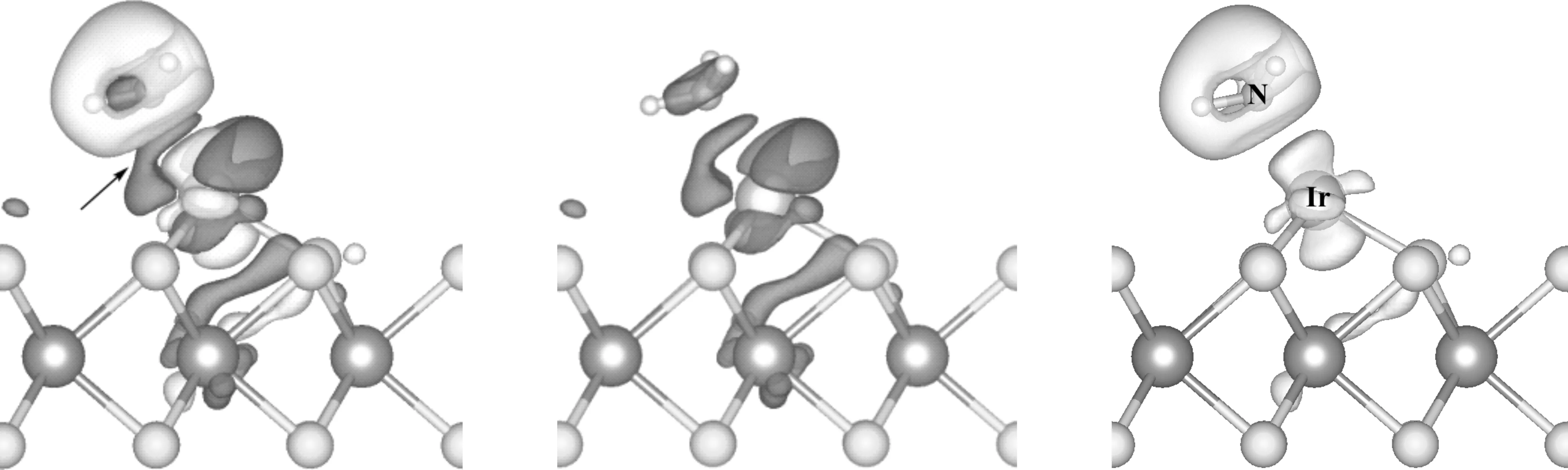
图3 电荷密度差分图Fig.3 The charge density difference diagram
为从微观上理解NH3在Ir1/MoS2体系上的吸附情况,我们计算了NH3在该催化剂表面上吸附前后的态密度DOS(即Density of Ststes(states/eV),计算结果如图4、图5所示,图中均以费米能级为0eV.由图4可知,NH3在Ir1/MoS2面上吸附后,各电子峰向能量更低的方向移动,显示整个体系更稳定,其中2a1和1e衍生态的DOS峰结构分布基本没有变化,说明了NH3分子的2a1和1e轨道与金属原子之间没有发生相互作用.而3a1衍生态的DOS峰与气态相比明显变得平坦且宽,且峰的位置移到费米能级以下,显示3a1轨道与体系Ir1/MoS2之间有很强的相互作用.
为了探究NH3分子的3a1轨道与体系的哪些轨道相互作用,将NH3/Ir1/MoS2体系的DOS分别投影到N原子和吸附位点的Ir原子上,如图5所示,可以发现3a1轨道与Ir原子的5S、5dyz、5dz2、5dxz轨道的峰有不同程度的重叠,其中5S、5dyz两轨道作用较为明显,说明NH3与体系Ir1/MoS2的相互作用主要是通过3a1轨道与Ir原子的5S、5dyz轨道相互杂化产生的.
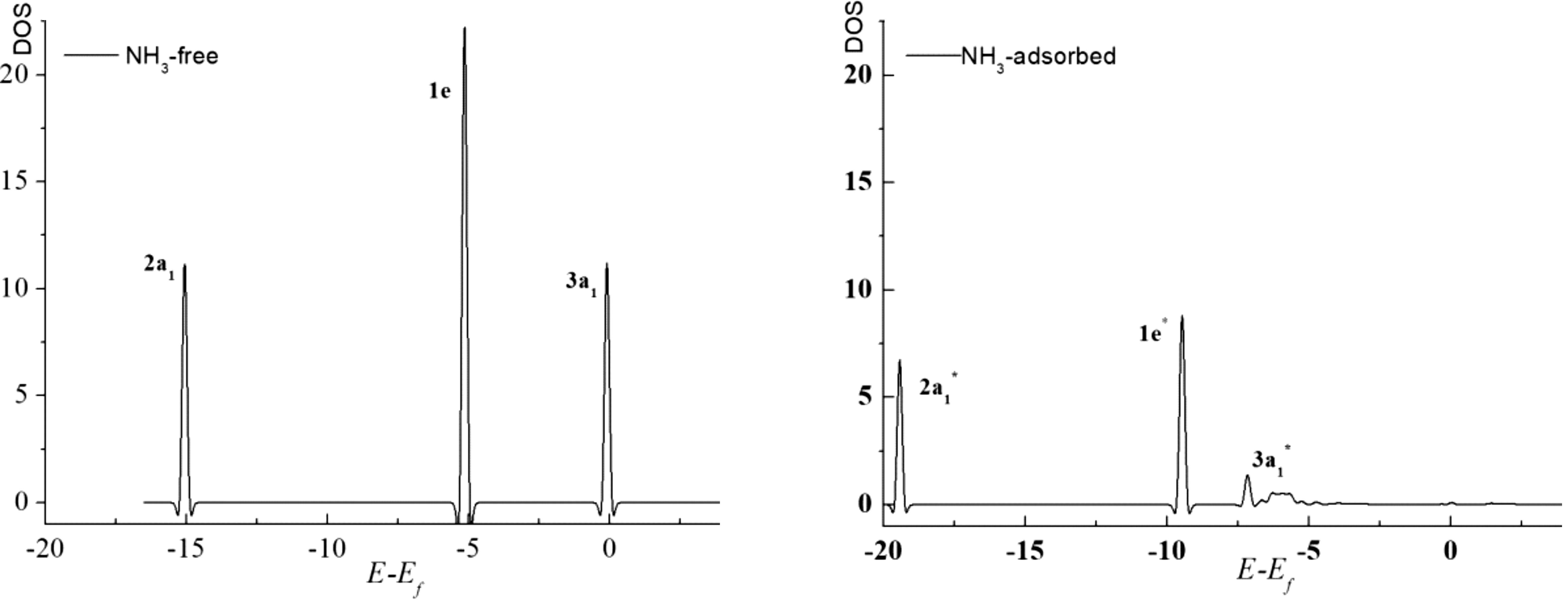
图4 NH3分子在Ir1/MoS2上吸附前后投影到N原子的DOS图Fig.4 The DOS diagram of NH3projected to N atom before and after adsorption on Ir1/MoS2
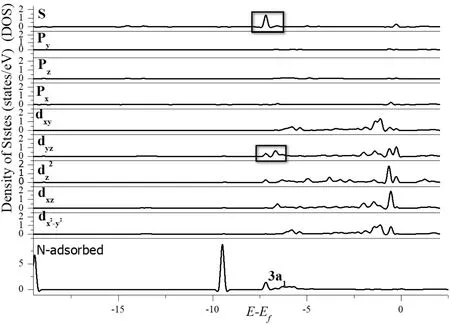
图5 NH3/Ir1/MoS2体系的部分态密度图Fig.5 The partial density of states diagram of NH3/Ir1/MoS2system
3 结论与讨论
利用第一性原理对NH3在单原子催化剂Ir1/MoS2上的吸附机理进行了研究.结果表明,单个Ir原子吸附在单层MoS2表面的三重空位(hcp)时整个体系稳定;NH3分子通过N原子和MoS2表面上的Ir原子相连形成体系Ir1/MoS2,最稳定位置的吸附能为1.63 eV;电子结构分析显示NH3的3a1轨道与Ir原子的5S、5dyz、5dz2、5dxz轨道的峰有不同程度的重叠,而其中 5S、5dyz两轨道起着主要作用.
猜你喜欢
证券市场周刊(2024年13期)2024-04-16 04:33:35
物理通报(2024年4期)2024-04-09 12:41:28
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
贵金属(2021年1期)2021-07-26 00:39:20
贵金属(2021年1期)2021-07-26 00:39:20
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26 00:58:18
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
新高考·高一物理(2015年6期)2015-09-28 20:10:57
