
原发性胆汁性肝硬化合并局灶节段性肾小球硬化一例并文献复习
2021-03-06 08:14:00孙婧黄华冰王歆妤梅小斌
临床肾脏病杂志 2021年2期
孙婧 黄华冰 王歆妤 梅小斌
1海军军医大学附属长海医院肾内科,上海 200433;2海军军医大学附属长海医院消化内科,上海 200433;3海军军医大学附属长海医院感染科,上海 200433
病例资料
患者,女,50岁。2018年2月15日患者无明显诱因出现双下肢及颜面水肿,至当地镇医院就诊,多次查尿蛋白均为(2+),遂于2018年2月28日至启东市人民医院就诊,查血肌酐(serum creatinine,Scr)48 μmol/L,白蛋白(albumin,Alb)25 g/L,碱性磷酸酶(alkaline phosphatase,ALP)1073 U/L,谷氨酰转肽酶(γ-glutamyl transpeptadase,γ-GT)1304 U/L,丙氨酸转氨酶(alanine aminotransferase,ALT)144 U/L,冬氨酸氨基转移酶(aspartate aminotransferase,AST)200 U/L,总胆固醇(total cholesterol,TC)17.09 mmol/L,乳酸脱氢酶(lactate dehydrogenase,LDH)13.33 U/L;乙肝两对半及丙肝标志物阴性;自身免疫指标:抗核抗体(+),抗线粒体抗体(antimitochondrial antibody,AMA)(+)。因患者不明原因肝损伤及血脂升高,建议转至上级医院。患者遂于2018年3月13日就诊我院。体格检查:血压 123/76 mmHg(1 mmHg=0.133 kPa),心脏各瓣膜区未闻及病理性杂音,双肺呼吸音清,未闻及干湿啰音,侧卧位可触及脾脏肋下2横指。双下肢轻度凹陷性水肿。实验室检查:24 h尿蛋白定量3.57 g;血生化检查结果示:Scr 53 μmol/L,Alb 27 g/L,球蛋白(glubulin,Glb)44 g/L,总胆红素(total bilirubin,Tbil)23.2 μmol/L,ALP 742 U/L,γ-GT 726 U/L,ALT 113 U/L,AST 114 U/L,TC 15.84 mmol/L,三酰甘油(triglyceride,TG)4.12 mmol/L,LDH 12.6 U/L;免疫球蛋白示:IgG 18.3 g/L,IgM 12.8 g/L,自身免疫抗体全套:抗核抗体(antinuclear antibody,ANA)胞浆型(+),AMA(+),抗线粒体抗体M2亚型AMA-M2(++)。心电图、胸部X线检查结果均无异常。腹部超声及CT增强扫描结果示肝右叶斑点钙化灶及小囊肿,双肾斑点钙化灶及多发囊肿,脾大。肾活检病理:肾小球数21个,其中3个肾小球球性硬化。少数肾小球球囊周纤维化,其内毛细血管襻缺血、皱缩。肾小管上皮细胞颗粒及空泡变性,管腔内可见蛋白管型,小灶状萎缩(萎缩面积约5%),肾间质灶状炎症细胞浸润,细动脉节段性玻璃样变,小动脉管壁增厚,管腔缩小。石蜡切片免疫荧光染色结果显示IgG(-),IgM(+),IgA(++),补体C3(-),补体C1q(-),沿系膜区颗粒样沉积。电镜下检测到1个肾小球。毛细血管内皮细胞明显空泡变性,管腔内可见红细胞聚集,毛细血管襻开放。肾小囊壁层无明显增厚,壁层细胞空泡变性,无明显增生。肾小球基底膜:无明显增厚,厚度约210~410 nm,节段性皱缩。脏层上皮细胞:上皮细胞肿胀,空泡变性。足突弥漫融合,有微绒毛变。系膜区:系膜细胞和基质增生,少数系膜区可见少量电子致密物沉积。肾小管-间质:肾小管上皮细胞空泡变性。病理诊断:符合足细胞病伴IgA沉积,可见肾小球硬化及肾小管萎缩,高度考虑局灶节段性肾小球硬化(focal segmental glomurular sclerosis,FSGS)(图1)。根据患者临床表现及检查结果,诊断(1)肾病综合征 局灶节段性肾小球硬化,(2) 原发性胆汁性肝硬化。予熊去氧胆酸(250 mg每次,每日2次口服)联合多烯磷脂酰胆碱(456 mg每次,每日2次口服)利胆保肝、百令胶囊(2 g每次,每日3次口服)护肾等对症支持治疗6周后,患者水肿消退,复查尿蛋白1+,24 h尿蛋白定量0.48 g,血液生化检查结果:Scr 43 μmol/L,Alb 34 g/L,Glb 41 g/L,Tbil 16.6 μmol/L,ALP 457 U/L,γ-GT 386 U/L,ALT 79 U/L,AST 71 U/L,TC 13.21 mmol/L,TG 1.85 mmol/L,LDH 9.39 U/L。免疫球蛋白示:IgG 17 g/L,IgM 6.7 g/L,自身免疫全套:ANA胞浆型(+),AMA(+),AMA-M2(++)。
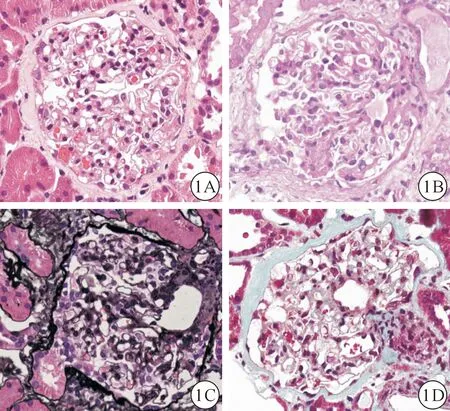
图1 肾穿刺病理图片(光镜400×) 图1A.HE染色;图1B.PAS染色;图1C.PASM染色;图1D.Masson染色
讨 论
原发性胆汁性肝硬化又名原发性胆汁性胆管炎(primary biliary cirrhosis,PBC)其本质是一种免疫介导的中小型肝内胆管破坏为特点的慢性进行性胆汁淤积性肝脏疾病,最终可发展为肝硬化。PBC发病与异常自身免疫有关,常合并有类风湿关节炎、慢性甲状腺炎、干燥综合征、系统性硬化症、系统性红斑狼疮、自身免疫性肝炎等疾病,既往文献合并肾脏损害的报道较少,本文报道PBC合并FSGS一例,并通过文献复习分析疾病发病机制特点及指导临床诊治。
PBC多见于50岁以上中老年女性,有研究显示男女比例为1∶9[1],病死率在肝硬化中占0.6%~2.0%,占国外成人肝移植指征的第2位[2]。其临床常表现为乏力(约见于40%~80%的患者)、皮肤瘙痒(约见于20%~70%的患者)[3],实验室检查主要为ALP及γ-GT升高,体液免疫异常,特征为IgM升高,且在PBC早期阶段,伴IgA及IgG正常或增高,慢性阶段仅血清IgM明显升高。抗线粒体抗体AMA阴性的PBC患者IgM不升高而IgG升高。免疫学指标是最重要的诊断手段,抗线粒体抗体AMA阳性,尤其AMA-M2高度阳性敏感度和特异度高,均超过95%,常伴有ANA或其他自身抗体阳性。且有报道指出,90%~95%的PBC患者可能在发病前几年就存在AMA,此外,90%的无症状AMA阳性患者的组织学特征与PBC一致[4]。治疗上,熊去氧胆酸是目前唯一推荐用于治疗PBC的一线药物[5]。有研究发现熊去氧胆酸在改善肝功能同时,可引起AMA滴度降低、血浆免疫球蛋白下降及一些免疫标志物的减少。熊去氧胆酸可取代内源性胆汁酸,减弱内源性胆汁酸的肝细胞毒性,还可以作为免疫调节剂降低黏附分子、HLA抗原在肝细胞和胆管上皮细胞的异常表达。PBC的诊断一般基于以下至少两个标准,(1)胆汁淤积伴碱性磷酸酶活性升高的生化证据;(2)AMA阳性。如果AMA阴性,则存在其他PBC特异性自身抗体,包括抗sp100抗体或抗gp210抗体。(3)组织学证据显示非化脓性胆管炎和小叶间胆管破坏。鉴别诊断包括胆汁淤积药物反应、胆道梗阻、结节病、自身免疫性肝炎(autoimmune hepatitis,AIH)和原发性硬化性胆管炎[6]。本例患者为中年女性,起病隐匿;生化提示ALP、GGT显著升高,ALT、AST为正常上限的2~3倍;免疫学检查提示免疫球蛋白IgM升高明显,AMA、AMA-M2及ANA均阳性,满足PBC诊断标准。
有15%的PBC患者有遗传风险,环境风险已经被证实[7],几项大型病例对照队列研究发现与尿路感染、生殖激素替代、指甲油和吸烟史有关,遗传和环境效应之间相互作用促进疾病的发生[6]。至今,PBC的具体发病机制仍不明确,但研究证实其与异常自身免疫有关。研究表明PBC的发生与T细胞的异常活化有关。PBC 的发病机制主要是自身反应性T细胞的过度免疫反应。PBC患者受损的门管区存在大量的CD4+T淋巴细胞浸润[8],CD4+T淋巴细胞尤其是辅助性T淋巴细胞Th1和Thl7可能参与了PBC的炎症反应过程,有研究显示在干燥综合征合并PBC患者外周血中TH17/调节性T淋巴细胞水平差异性失衡,Th17细胞很大程度上参与了PBC的发病[9]。PBC患者常合并有类风湿关节炎、慢性甲状腺炎、干燥综合征、系统性硬化症、系统性红斑狼疮、自身免疫性肝炎等疾病,既往文献合并肾脏损害的报道较少,Lino等[10]报道了4例PBC合并肾小管间质性肾炎患者,认为异常抗原表达于肾小管上皮细胞,通过自身免疫T细胞反应介导肾间质损害造成肾小管间质性肾炎,并且三分之一的晚期PBC患者被诊断为远端肾小管酸中毒,但通常没有临床意义。PBC合并NS较为罕见,据报道,至今PBC合并最多的NS病理类型为膜性肾病,共有14例[11];合并微小病变1例[12];合并系膜增生性肾小球肾炎2例[13-14];本例为首次PBC合并FSGS报道。
原发性FSGS占黑人肾病综合征(nephrotic syndrome,NS)的50%以上,是黑人原发性NS最常见的病理类型[15]。在中国,FSGS占终末期肾病的3.2%~5.8%[15]。临床和实验研究证实,足细胞结构和功能部分或全部性破坏是FSGS起病和进展中的关键因素[16],因此FSGS被认为是足细胞病,发病与不可逆的足细胞应力导致足细胞脱离或凋亡有关。其他参与的因素有机械剪切力、内质网应激、细胞黏附减弱、促凋亡途径的激活、基因突变、免疫异常密切相关,常用治疗药物为激素和免疫抑制剂。本例患者以双下肢及颜面部水肿起病,同时有大量蛋白尿、低蛋白血症、高脂血症,肾活体组织检查结果符合足细胞病伴IgA沉积,可见肾小球硬化及肾小管萎缩,高度考虑FSGS。综上,肾病综合征(局灶节段性肾小球硬化)诊断明确。
目前多数观点认为,FSGS发病机制中足细胞损伤是始发因素,而后出现肾小球其他损伤,如基底膜塌陷、球囊粘连、细胞增生、泡沫细胞及玻璃样变性等[16]。FSGS中常有IgM沉积,但一般认为系血浆蛋白非特异性沉积,而不是体液免疫反应的证据。有研究发现FSGS肾小球硬化区有高强度C4d表达,尤其是在血管袢呈不均匀粗线状沉积,这一证据表明补体经典途径激活参与FSGS的发病机制[17]。微小病变(minimal change disease,MCD)和FSGS都是足细胞病变,均表现为足突融合和肾性蛋白尿,在一项儿童MCD研究中发现T细胞白细胞介素-13(interleukin -13,IL-13)表达增加,用IL-13过度表达可模拟出大鼠MCD模型,表明足细胞是先天免疫系统的一个成员[18]。有研究发现Th17在FSGS外周血的比例很高,表明的CD4+T淋巴细胞参与了FSGS的发生发展[19]。综上所述,CD4+T淋巴细胞参与PBC及FSGS发病机制,提示PBC与FSGS合并存在可能。
Bindi等[20]的研究发现PBC合并膜性肾病患者存在肾组织上皮下AMA沉积;Sato等[11]统计了14例PBC合并膜性肾病患者病例,发现血清IgM升高的患者中,60%存在肾小球基底膜IgM沉积。本例患者也存在血清IgM升高及肾小球系膜区IgM沉积,提示PBC与FSGS发病可能存在共同的免疫机制,但仍需要进一步研究以明确。
PBC+FSGS可能为以下两种情况:(1)PBC与FSGS并存;(2)PBC合并有继发FSGS。继发性FSGS多由肾小球高压力、高灌注引起的结构和功能的代偿性变化,可继发于高血压、糖尿病、肥胖、紫绀型先天性心脏病或伴肾实质减少的疾病,如肾发育不良、孤立肾、反流性肾病、肾脏切除术后等,病毒感染、药物因素亦可导致FSGS,继发性FSGS蛋白尿的程度相对较轻,很多患者表现为非肾病范围蛋白尿,低蛋白血症的发生率亦相对较低。该例患者既往无高血压、糖尿病等慢性疾病,无肾脏手术史,无感染、用药史,体重指数在正常范围,继发性FSGS可能性小。而综合上述发病机制,PBC和FSGS可能存在共同发病机制,且本例患者未经激素及免疫抑制剂治疗,经过熊去氧胆酸的治疗,肝功能改善,NS自发缓解,提示PBC与FSGS合并存在可能。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
沉积与特提斯地质(2019年1期)2019-07-16 08:41:30
沉积与特提斯地质(2018年4期)2018-07-16 08:27:30
中外医疗(2016年15期)2016-12-01 04:25:52
南方文学(2016年4期)2016-06-12 19:58:50
中外医疗(2015年11期)2016-01-04 03:58:45
中国塑料(2015年11期)2015-10-14 01:14:16
医学研究杂志(2015年9期)2015-07-01 17:28:00
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
西南军医(2015年6期)2015-01-23 01:25:49
