
毛竹水解渣木质素氢解制备芳香化学品
2021-03-05 08:22屈一新王际东
林产化学与工业 2021年1期
程 毅,屈一新,杨 昇,庄 抗,王际东*
(1.北京化工大学 化学工程学院,北京 100029;2.中国林业科学研究院木材工业研究所,北京 100091)
生物炼制是以生物质为原料,通过物理、化学或生物手段,获得生物基燃料、材料或化学品的过程。木质纤维是一种储量丰富且可再生的生物质资源,主要由纤维素、半纤维素和木质素组成[1]。典型的木质纤维生物炼制过程是先将原料的目标组分分离,再进一步转化[2-3]。过程中产生的副产物往往被加工成低附加值产品或直接燃烧,难以得到高值化利用。优化纤维素、半纤维素和木质素的分离工艺,开发新型反应体系,实现全组分炼制,近年来受到越来越多学者的关注[4]。木质素可以通过化学或物理方法进行分离,进而实现各组分转化。常见的木质素的化学分离方法包括过氧化氢法、有机溶剂法、碱法和亚硫酸盐法等。于海龙[5]在对工业纤维渣脱木质素研究中发现:相同条件下,过氧化氢法分离糠醛渣中的木质素,效果优于含半纤维素的甘蔗渣;而有机溶剂法的结果正相反。这说明解除了半纤维素屏障的木质纤维,采用过氧化氢法分离木质素可以取得很好的效果。过氧化氢法分离木质素的过程中,脂肪族羟基被氧化成羰基或醛基,降低了木质素中氧醚键的解离能[6]。因此,过氧化氢法从戊糖水解渣中分离的木质素在化学品转化方面拥有巨大潜力。过渡金属催化剂,如Pd/C、Ru/C或Ni/C等,在供氢溶剂(甲醇、乙醇或四氢呋喃等)中转化芳香化学品被证明是木质素转化的一种有效方式[7-10]。为了防止降解产物发生再缩合,溶剂会采用供氢溶剂和水的混合体系,如甲醇/水或乙醇/水[11-13],提高目标产品的得率。本研究以毛竹提取半纤维素后的水解渣为原料,分别利用过氧化氢法和甲醇法提取木质素,以甲醇/水作为反应溶剂和氢源,Pd/C为催化剂,对两种木质素进行氢解,获得芳香化学品,使用GC-MS、FT-IR和GPC等手段对原料和氢解得到的产品结构进行分析,并据此对毛竹水解渣木质素氢解过程进行探索。
1 实 验
1.1 材料、试剂与仪器
原料为三年生毛竹(Phyllostachysheterocyclacv.Pubescens),由国际竹藤中心提供。将竹节粉碎后筛分得到粒径小于0.425 mm的样品,平均含水率为9.3%,备用。绝干原料中纤维素、半纤维素和木质素质量分数分别为46%、24.9%和25%。
催化氢解所用的催化剂为Pd/C,Pd负载量为5%,美国Sigma-Aldrich公司。甲醇,分析纯;FeCl3·6H2O,试剂纯;四氢呋喃、甲醇和溴化钾,均为光谱纯。
Waters 8795e高效液相色谱(HPLC)仪,带有示差检测器(RID),美国Waters公司;色谱柱Aminex HPX-87P column (300 mm×7.8 mm),美国Bio-Rad公司;GC-2010 Plus气相色谱-质谱联用(GC-MS)仪,KB-WAX分离柱(0.25 mm×30 m×0.25 μm),日本岛津公司;Tensor 27傅里叶变换红外光谱(FT-IR)仪,德国布鲁克公司;Waters 7890e凝胶渗透色谱(GPC),水相色谱柱PWxL Guard Column,TSKgel G3000PWxL和TSKgel G5000PWxL串联,日本TOSOH公司;PLgel-Mixed-D四氢呋喃相色谱柱(300 mm×7.5 mm),美国Agilent公司。
1.2 毛竹水解渣的制备
参考文献[14]的方法制备毛竹水解渣。取13.23 g原料(绝干物12 g)加入到200 mL高压釜中,再加入4 g FeCl3·6H2O和120 mL水。搅拌下加热至170 ℃保持35 min。反应结束后,将反应釜在冷水中急速冷却至室温,将混合物过滤分离。所得固体经水洗至水洗液为无色,在105 ℃烘箱中绝干4 h,即得到毛竹水解渣。
1.3 木质素的提取制备
1.3.1过氧化氢法 参考文献[5]的方法,取0.75 g毛竹水解渣加入到25 mL聚四氟乙烯水热釜中,加入14 mL pH值10.5的NaOH溶液(质量分数0.01%)和1 mL H2O2溶液(质量分数30%),将釜盖迅速关闭拧紧;放入恒温水浴锅中,在磁力搅拌下于80 ℃反应4 h;然后,将反应釜在冷水中急速冷却至室温,过夜。反应结束后,过滤,将得到的处理液经真空旋转蒸发浓缩至5 mL,处理液从浅黄色变成深棕色,将该深棕色液体逐滴加入到50 mL 95%乙醇中,所得悬浊液在室温下静置过夜后过滤,滤饼在空气中风干,即可得到过氧化氢木质素。
1.3.2甲醇法 参考文献[5]的方法,取1.5 g毛竹水解渣加入到25 mL聚四氟乙烯水热釜中,再加入7.5 mL的甲醇和7.5 mL pH值10.5的NaOH水溶液;反应釜密封后,恒温油浴中加热到140 ℃,磁力搅拌3 h。反应结束后,将反应釜在冷水中急速冷却至室温,经过滤分离,得到含有甲醇木质素的处理液。将该处理液经真空旋转蒸发浓缩至5 mL,然后滴加到50 mL pH值2的盐酸(体积分数0.3%)中,所得悬浊液在室温下静置过夜后过滤分离,将滤饼用去离子水洗至中性,冷冻干燥48 h,干燥后的滤饼,即为甲醇木质素。
1.4 木质素氢解制芳香化学品
将0.5 g的木质素、0.05 g Pd/C催化剂和10 mL甲醇/水(体积比1 ∶1)溶液加入到水热釜中,密封后放入油浴中加热至所需的反应温度,反应一定时间后,将水热釜迅速冷却至室温,然后过滤分离釜内混合物。滤饼用反应溶剂洗至无色,烘干后称质量,计算固体残渣质量。
滤液为木质素的氢解产物及溶剂。取1 mL滤液与等体积含有十二烷内标物的二氯甲烷混合,萃取,取下层萃取液用气相色谱测定氢解产物中芳香族单体的含量。剩余滤液在35 ℃下真空干燥4 h,除去溶剂,所得油状物质称质量后采用气相色谱分析单体含量。可溶物得率和芳香族单体产率计算公式如下:
式中:ys—可溶物得率,%;mr—固体残渣质量,g;mL—起始木质素加入量,g;ya—芳香族单体产率,%;ma—芳香族单体质量,g。
1.5 分析与表征
1.5.1HPLC分析 毛竹水解渣及提取到的木质素的组分分析方法参考文献[5]。准确称量0.3 g绝干样品,置于耐压瓶中,加入3 mL 72%的硫酸,搅拌均匀,置于30 ℃恒温水浴中反应1 h。反应得到的固体残渣为Klason木质素及灰分,上清液为多糖水解产物和少量酸溶木质素。将固体残渣在550 ℃马弗炉中煅烧4 h,煅烧后得到灰分质量,除去灰分后的固体质量即为木质素质量。上清液用碳酸钙中和后离心,然后采用高效液相色谱仪检测上清液组成,流动相为纯水,流速0.6 mL/min,柱温85 ℃,检测器温度35 ℃,进样量10 μL。
1.5.2GC-MS分析 采用GC-MS仪进行分析,FID为检测器。以高纯氮气为载气,正十二烷作为内标,柱箱的初始温度为50 ℃,以10 ℃/min的升温速率升温至250 ℃并保持5 min。产物首先使用GC-MS分析,所得图谱经过与NIST数据库比对后进行定性。
1.5.3FT-IR分析 在FT-IR仪上进行分析。将样品与溴化钾按照质量比1 ∶100混合磨碎并制成透明薄片,以纯溴化钾压片,在波数500~4000 cm-1,分辨率为4 cm-1的条件下进行分析。
1.5.4GPC分析 木质素及产品的相对分子质量测定采用凝胶色谱(GPC)法。过氧化氢木质素相对分子质量测定参考文献[15]报道的方法,将5 mg样品溶解在5 mL纯水中,溶解后液体采用水系滤膜过滤,使用纯水作为流动相,流速为0.5 mL/min,进样量50 μL,检测器为示差折光检测器,以已知相对分子质量的葡聚糖为标准品进行标定。甲醇木质素及降解产物的相对分子质量测定参考文献[16]报道的方法,将5 mg样品溶解在5 mL色谱纯四氢呋喃中,溶解后液体采用有机系滤膜过滤,四氢呋喃作为流动相,流速为1 mL/min,进样量50 μL,检测器为示差折光检测器,以已知相对分子质量的聚苯乙烯样品为标准品进行标定。
2 结果与讨论
2.1 木质素的提取
毛竹水解渣以木质素和纤维素为主,含有少量的半纤维素。水解渣得率68.2%,水解液中木糖和阿拉伯糖的得率分别为原料总质量的20.5%和1.4%,由此计算得水解渣的纤维素、半纤维素和木质素质量分数分别为65.9%、4.9%和29.2%。以毛竹水解渣为原料,采用过氧化氢法提取木质素,木质素提取率为97.9%。醇析后得到的过氧化氢木质素占毛竹水解渣总质量的27.2%,其中木质素的质量分数为91.6%,其余为多糖组分。采用甲醇法提取木质素,木质素提取率为76.4%。酸析后得到的甲醇木质素占毛竹水解渣总质量的26.7%,其中木质素的质量分数为77.1%,葡聚糖质量分数22.3%,灰分质量分数0.6%。从木质素提取率和木质素产品纯度分析,过氧化氢法优于甲醇法。
2.2 木质素氢解工艺条件探讨
2.2.1过氧化氢木质素氢解产物分布 表1给出了不同反应温度和反应时间条件下,过氧化氢木质素转化得到的芳香族物质的分析结果。对过氧化氢木质素氢解转化的产物做进一步分析,可以将其分为两类:对位为脂肪族含氧取代的愈创木酚衍生物1~4(第I类)和对位为烷烃取代的愈创木酚衍生物及愈创木酚5~8(第II类)。

表1 反应条件对过氧化氢木质素氢解的影响Table 1 Effect of reaction conditions on hydrogenolysis of hydrogen peroxide lignin
表1还给出了这两类产物收率随反应温度和反应时间变化的趋势。可以看出,随着氢解反应时间的延长,第I类产物的总收率先增加后降低,反应时间大于60 min后明显降低。第I类产物的总收率从60 min时的7.44%降至120 min时的1.11%。这类产物总收率的降低是由每种产物收率降低的叠加导致的。其中,降低最明显的物质为产物3,从3.84%降至0.26%。对于第II类物质,产物5和6在反应时间为60 min时产率最高。随反应时间延长,产物7的收率逐渐升高,而产物8的收率在逐渐降低。随着时间的延长,可溶物的收率呈现降低的趋势,当反应时间大于90 min后降低最为明显。不溶物部分可能含有未反应的木质素和木质素氢解产物二次反应生成的不溶性固体。此外,在220 ℃、反应时间为30~90 min时,木质素氢解产物二次反应很少,但反应时间大于90 min后会有明显的增加。由此可知,氢解反应时间对于过氧化氢木质素的转化及产物分布均有较大的影响,延长反应时间使芳香族单体物质的总收率降低,而其中一些物质的收率可能会出现极大值,在反应时间大于90 min后,芳香族单体物质的二次反应会明显增加。
表1还给出了反应时间为60 min时,反应温度对产物收率的影响。随着反应温度的提高,第I类产物的收率呈现先升高后降低的趋势。第II类产物的总收率逐渐提高,其中产物5和6的收率随着反应温度升高而提高最为明显,当反应温度为260 ℃时,产物5和6的收率分别为4.59%和2.69%。产物7和8的收率则随着温度的提高呈现先升高后降低的趋势。随着温度升高,木质素转化的可溶物的收率在220 ℃时最高。这一现象可能意味着,温度低于220 ℃时有少量的木质素没有转化,而温度高于220 ℃ 时,氢解产物的二次反应会加剧。综合上述的结果可以认为,以Pd/C作为催化剂,以甲醇/水为反应溶剂,氢解过氧化氢木质素时,反应温度220 ℃,反应时间60 min,氢解产物得率最高。该反应条件将进一步用于甲醇木质素降解,考察其转化效率。
2.2.2两种木质素氢解产物对比 生物炼制过程,有机溶剂木质素(甲醇、乙醇或二氧六环木质素等)被认为是除磨木木质素外,最接近原生结构的木质素。在很多模型化合物转化的研究中,大多会采用有机溶剂木质素做进一步验证[17]。因此,本研究对比了相同反应条件(反应温度220 ℃,反应时间60 min)下甲醇木质素的转化情况。
甲醇木质素在相同反应条件下,转化结果与过氧化氢木质素有明显差别。甲醇木质素氢解转化的芳香单体产物包括4-(4-羟基苯基)-2-丁酮0.95%,4-(4-羟基-3-甲氧基苯基)-2-丁酮2.00%,3-(4-羟基-3-甲氧基苯基)-1-丙醇1.13%和3-(4-羟基-3,5-二甲氧基苯基)-1-丙醇3.05%,单体总产率为7.13%,可溶物收率为96.8%。过氧化氢木质素的氢解产物以愈创木基型(G型)为主,酚羟基对位的脂肪族官能团的碳数为0~3;甲醇木质素的氢解产物包括愈创木基型(G型),紫丁香基型(S型)和对羟苯基型(H型)3种,酚羟基对位的脂肪族官能团碳的数目为3或4。除了产物种类的明显差别外,两种木质素转化得到的芳香化学品的含量也存在较大差别。在220 ℃反应60 min 后,过氧化氢木质素氢解得到的芳香族物质总量为14.84%,而甲醇木质素氢解得到的芳香族物质总量仅为7.10%。过氧化氢木质素氢解得到的香草酮(3)、香草醛(4)分别为3.84%和1.98%,这两种芳香化学品都是比较重要的香料或有机合成中间体。
2.3 木质素氢解产物分析
2.3.1相对分子质量分布 木质素是一种天然高分子物质,其氢解产物中除上述的芳香族单体物质外也含未降解的高分子物质。因此,也需要对这些高分子物质的性质进行分析,以便对于木质素及其降解产物的性质有较全面的了解。
图1给出了过氧化氢木质素、甲醇木质素及其降解产物的GPC图谱。过氧化氢木质素相对分子质量较大,其数均相对分子质量(Mn)为44 529,氢解后降至1 397,表明其发生了很明显的降解;甲醇木质素的数均相对分子质量为1 581,氢解后降至1 183,降低并不明显。
从分散系数(Mw/Mn)可以看出,过氧化氢木质素的分散系数为1.06,氢解后分散系数为1.46,说明氢解产生了更多的低相对分子质量产物,使相对分子质量分布范围变广。与之相反,甲醇木质素的分散系数在氢解前后分别为1.39和1.16,说明降解后甲醇木质素的相对分子质量分布范围变得更为集中。两种木质素相对分子质量分布的结果表明:过氧化氢木质素在氢解过程中比甲醇木质素更容易发生降解。
2.3.2FT-IR分析 图2给出过氧化氢木质素、甲醇木质素及降解产物的FT-IR谱图。
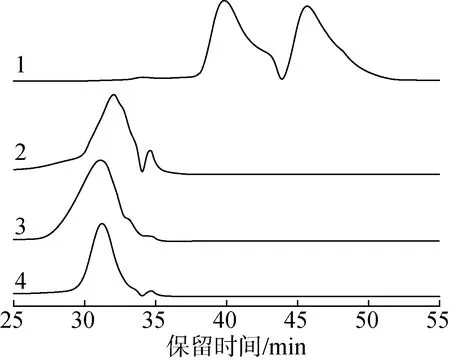

1.过氧化氢木质素hydrogen peroxide lignin;2.过氧化氢木质素降解产物degradation product of hydrogen peroxide lignin;3.甲醇木质素methanol lignin;4.甲醇木质素降解产物degradation product of methanol lignin
信号归属参考Wang等的研究[16]。从图中可以看到,过氧化氢木质素降解后指纹区(1000~2000 cm-1)的红外吸收信号数量增多,吸收峰位置发生了显著的变化;甲醇木质素在降解后红外吸收信号的降低相对较小,且吸收峰的位置没有明显的变化。在3403 cm-1处的宽吸收峰为O—H伸缩振动,对应木质素中脂肪族基团;2939 和1458cm-1处的吸收峰为甲基和亚甲基C—H伸缩振动;2841和1427 cm-1处的吸收峰为甲氧基中C—H伸缩和变形振动。
从图2可看到,过氧化氢木质素在1458 cm-1处的吸收峰明显强于甲醇木质素,该信号归属于甲基和亚甲基基团。此处的明显差异表明,过氧化氢提取木质素会破坏苯环结构,发生开环,使苯环上共轭的碳氢转变为非共轭的碳氢,从而提高脂肪族碳氢(如亚甲基)的相对含量,降低苯环信号的比例。

2.4 两种木质素的结构差异分析
对于过氧化氢木质素和甲醇木质素结构的差异,可以结合过氧化氢与木质素的反应机理(图3)进行解释。碱性条件下,H2O2分解成过氧酸根离子(HOO-),使脂肪族α-C和γ-C分别转变为过羟基和醛基,过羟基进一步脱氢氧根,变成氧负离子[18-19],此时木质素氧醚键断裂的解离能较低,更容易发生解聚。此外,过氧化氢还有可能氧化苯环,使其转变为苯醌甚至开环:苯环甲氧基在碱性条件下受到过氧根离子的攻击,连接上一个氧负离子和一个过氧基,该结构会进一步转化为一对邻位苯醌。如果继续有过氧根离子存在,邻位苯醌之间的C—C就会发生断裂,形成两个带有负电荷的酯键。
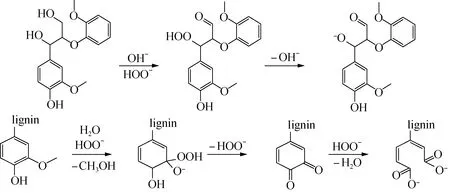
图3 过氧化氢氧化木质素机理Fig.3 Mechanisms of delignification process by alkaline hydrogen peroxide oxidation
除了官能团被修饰外,氧化还能够消除木质素分子内氢键,使大分子结构更加线性舒展。研究者利用密度泛函理论,结合半经验公式,对8个苯环组成的木质素的分子构象进行计算,受分子内的氢键影响,分子骨架呈折叠状态,宽度仅4.1 nm,任意两个原子之间宽度小于1.5 nm[15]。破坏氢键后得到相对线性的大分子木质素,有利于非均相催化反应的进行。
FT-IR分析说明过氧化氢木质素转化产品中芳香酸、酯和醛类多于甲醇木质素的转化产物,解释了氢解生成较多的第I类芳香族单体的原因。过氧化氢木质素反应前后指纹区信号的明显变化,说明氢解可以使被过氧化氢氧化的木质素还原成芳香族化合物。
甲醇木质素在该体系中转化为单体的含量相对于文献报道是较低的[3,17]。这可能是由于反应过程中,针对氧醚键的定向转化选择性不高,采用的催化剂和溶剂效率不足导致的[4,20]。但是本研究对两种木质素转化研究,采用的催化剂和溶剂均为木质素转化实验常用的反应体系。过氧化氢法提取毛竹水解渣中的木质素转化为芳香化学品的结果,证实其相比于甲醇法更具有优势。
3 结 论
3.1分别采用过氧化氢法和甲醇法,从毛竹水解渣中提取获得了两种木质素,将木质素氢解转化制芳香化学品,在反应温度220 ℃,反应时间1 h的优化条件下,过氧化氢木质素可以全部转化为可溶物,单体得率为14.85%;甲醇木质素的可溶物得率为96.8%,而单体得率仅为7.13%。
3.2对比了两种木质素的氢解产物,过氧化氢木质素转化产物以愈创木基型为主,酚羟基对位取代基团以羰基、醛基和烷烃取代基为主;FT-IR和GPC分析结果表明相比于甲醇木质素,过氧化氢木质素存在结构上的优势,更容易通过催化氢解,转化为芳香化学品。
猜你喜欢
氯碱工业(2022年3期)2022-11-25
造纸信息(2022年8期)2022-11-10
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
轮胎工业(2021年4期)2021-12-25
养殖与饲料(2021年5期)2021-11-30
意林·少年版(2020年13期)2020-08-02
东坡赤壁诗词(2020年2期)2020-06-04
新能源进展(2020年1期)2020-03-09
中国造纸(2019年6期)2019-09-10
阅读与作文(小学高年级版)(2019年3期)2019-04-20
