
白芷挥发油抗炎镇痛活性成分的虚拟筛选
2021-03-04 08:43汤建赵康琦张腾腾申金田闻崇炜
中医药信息 2021年2期
汤建,赵康琦,张腾腾,申金田,闻崇炜*
(1.亳州学院中药学院,安徽 亳州 236800;2.江苏大学药学院,江苏 镇江 212013)
中药白芷为伞形科植物白芷[Angelicadahurica(Fisch. ex Hoffm.)Benth. et Hook. f.]或杭白芷[Angelicadahurica(Fisch. ex Hoffm.)Benth. et Hook. f. var. formosana(Boiss.)Shan et Yuan]的干燥根,可用于解表散寒、祛风止痛、宣通鼻窍、燥湿止带、消肿排脓[1]。主流商品药材以川白芷、杭白芷、祁白芷、禹白芷为主[2]。
白芷的主要化学成分包括香豆素、挥发油以及甾醇类化合物等。其中挥发油的含量约占0.13%(以提取得率计),在药品、护肤品、香料等方面的开发和利用均具有较好的市场潜力。在一系列的活性研究中发现白芷挥发油具有良好的镇痛、抑制酪氨酸酶、抗氧化和抗过敏等生物活性[3-6]。其中抗炎镇痛活性与白芷药材的临床应用一致,但由于挥发性成分分离鉴定具有一定的难度,一般是作为挥发油部位(粗提取)来进行研究,尚未对白芷挥发油部位物质基础进行细致的药理活性研究,其作用机制尚不清楚。
目前,采用计算机软件进行分子对接(Molecular Docking),虚拟筛选活性部位的有效成分和潜在的相关蛋白,进而进行药理实验确证是一种高效的途径,这一策略已经成为一种有效地探索中药活性成分及其作用机制的途径。分子对接是利用计算机技术模拟分子的几何匹配和能量匹配,来寻找小分子配体(化合物)与大分子生物受体(靶点)之间活性位点的最佳结合模式的过程[7-8]。针对某一特定的靶点,利用相关软件对化合物进行对接,虚拟筛选出的化合物再进一步进行生物活性检测,最终确定活性成分,该技术在中药有效物质基础研究方面有着广泛的应用前景。目前常用的软件有AutoDock、MOE、Sybyl、eHiTS和Molegro Virtual Docker等[8]。
本研究中根据前期实验数据[6,9-14]选取κ阿片受体等18个蛋白为对接靶点蛋白,采用AutoDock Vina软件,与检索到的17个化合物进行分子对接,虚拟筛选白芷挥发油抗炎、镇痛活性成分,初步确定潜在的关键结合蛋白,为进一步探讨白芷抗炎镇痛机制提供一定的科学依据。
1 材料
本研究采用AutoDock Tools1.5.6和AutoDock Vina(美国The Scripps Research Institute),PyMOL(TM) 1.7.4.5 Edu(美国Schrodinger, LLC公司)和Discovery Studio 2016 client(美国BIOVIA公司)。
2 方法
2.1 白芷挥发油中化学成分的收集
检索国内外关于中药白芷的文献,共收集到含量较为丰富的17个小分子化合物(按结构类型和含量排序):十二烷醇(dodecanol,1)、十三烷醇(tridecanal,2)、十四烷醇(tetradecanol,3)、2-甲基-2-己醇(2-methyl-hexan-2-ol,4)、1-十六烯(1-hexadecene,5)、E-3-十二碳烯(E-3-eicosene,6)、E-9-十八碳烯(E-9-octadecene,7)、甲基环癸烷(methylcyclodecane,8)、1,4-环壬二烯(1,4-cyclononadiene,9)、环十二烷(cyclododecane,10)、环十四烷(cyclotetradecane,11)、2-羟基环十五烷酮(2-hydroxycyclopentadecanone,12)、4-萜烯醇(4-terpinenol,13)、1-α-松油醇(1-α-terpinenol,14)、α-蒎烯(α-pinene,15)、β-石竹烯(β-caryophyllene,16)、蛇麻烯(又称葎草烯或α-石竹烯,α-humulene,17)。主要为脂肪烃、脂肪醇、单萜、倍半萜等化合物(见图1,括号内为单一成分质量占挥发油部位百分比)。化合物三维结构经MM2力场进行最小能量优化后保存为mol2格式。AutoDock Tools1.5.6对小分子进行加氢,计算Gasteiger电荷,合并非极性氢,结果保存为pdb格式,再转化成pdbqt格式,以备AutoDock Vina进行分子对接。
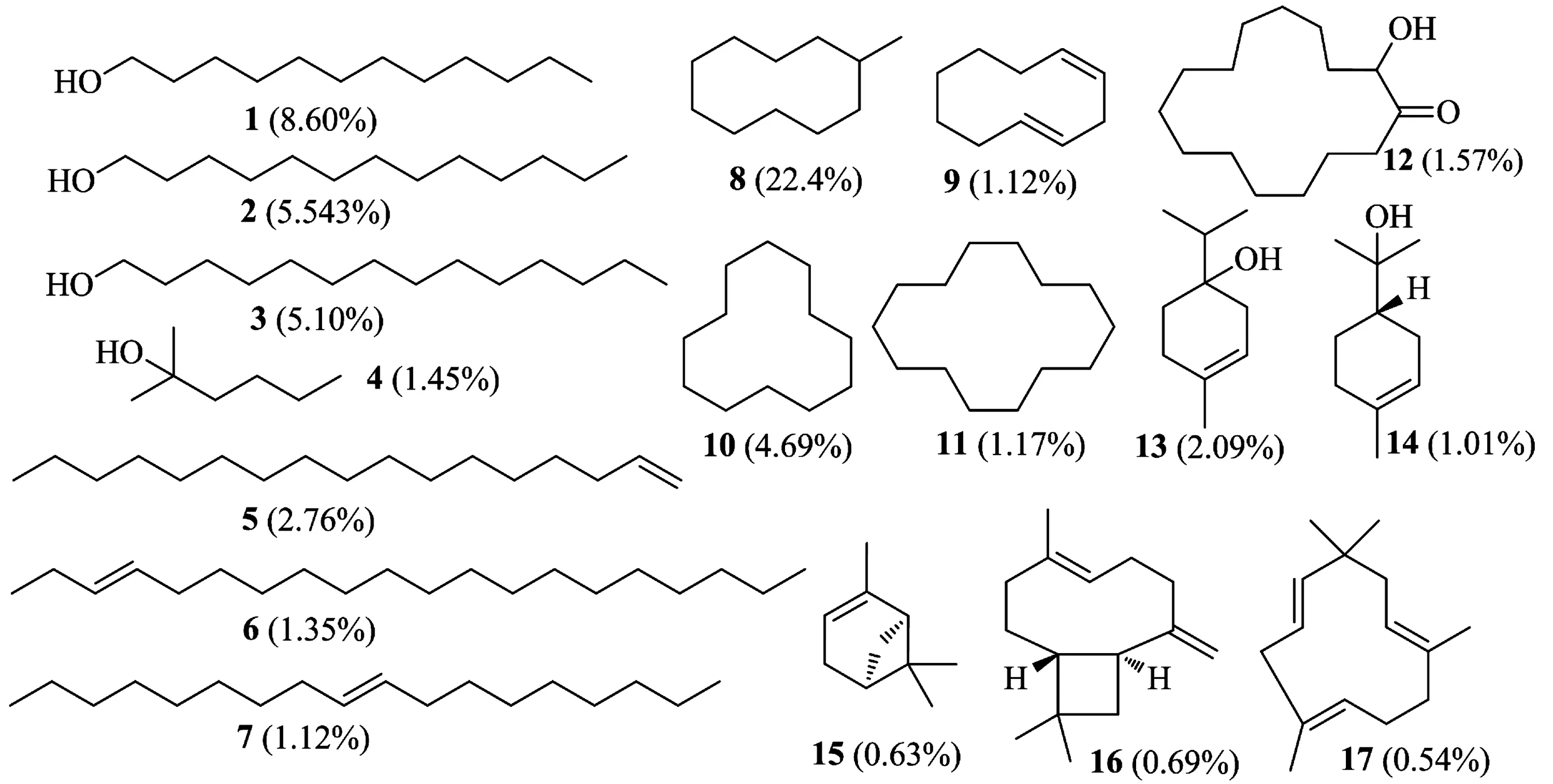
图1 白芷挥发油中的主要化学成分结构式(括号内为质量百分比)
2.2 靶标蛋白结构的获取和预处理
通过检索搜索PDB结构数据库(http://www.rcsb.org),获取18个靶点蛋白,见表1,将这些蛋白依次导入AutoDock Tools1.5.6中,删除原配体,水分子,加氢,保存为pdbqt格式,以备AutoDock Vina进行分子对接。

表1 抗炎镇痛相关靶点蛋白
2.3 分子对接
首先将靶点蛋白晶体复合物中配体取出,然后利用AutoDock Vina将配体分子与受体重新对接,其打分值为阈值。均方根偏差(RMSD)在2范围内,能够用于虚拟筛选。AutoDock Vina采用的是半柔性格点对接,指化合物对接蛋白质前要计算化合物中每种原子和受体结合区域之间的相互作用能量。对接过程中,对17个小分子化合物每个分别进行20次对接,其他参数均为缺省值。对接打分,得到17个化合物与18个靶点蛋白对接后的能量值,将能量值优于或接近该阈值的化合物认定为白芷挥发油中基于靶点的抗炎镇痛活性成分。
将蛋白导入AutoDock Tools1.5.6中,删除原配体和水分子,加氢,保存为pdbqt格式。以备Vina进行分子对接。将蛋白晶体复合物中配体取出,利用Vina将其与受体重新对接,其结合能分值为阈值。分子对接的中心的设置见表2,对接空间(Grid Box)设置为:30 mm×30 mm×30 mm,能量范围为4。利用Vina软件进行半柔性对接,得到对接后能量值。将能量值优于或接近阈值的化合物认定为中药白芷挥发油镇痛活性成分。
3 结果
3.1 分子对接的结果
从表2的对接打分值(结合能)可以看出,链状脂肪烃(醇)(1、2、4、5、6、7)在分子对接中所得的结合常数较环状脂肪烃及类似物(8、9、10、11、12)和萜类成分(13、14、15、16、17)低。其中打分值最低的短链脂肪烃(醇):1、2、4、5,其碳链长度为7~14。而碳链稍长的6 和7的结合力稍强一些。与链状脂肪烃(醇)比较,环状脂肪烃(醇),如8、9、10、11、12的结合力要明显提高,特别是化合物12,与TLR2(PDB ID:5D3I)结合常数为-8.3 kcal/mol,甚至超过了TLR2与原配体PCW的结合常数(-7.4 kcal/mol)。进一步比较发现,碳原子数为10~12的化合物8、9、10的结合力弱于碳原子数为14的化合物11和12。倍半萜化合物16和17对18种蛋白(基因)普遍具有较强的结合力,特别是对TLR2、κ阿片受体(PDB ID:4DJH)、5HT2AR(PDB ID:6A93)等蛋白,结合常数-8.0 kcal/mol左右。但单萜类化合物13、14、15的结合常数普遍较低,这与已有的4-萜烯醇(13)、松油醇(14)以及α-蒎烯(15)的良好抗炎活性的报道不尽一致[15]。在利用软件进行分子对接过程中,配体化合物除了应具有相关的官能团外还应具备一定的空间体积,以便于与所处位置的氨基酸残基有较强的结合,但单萜类化合物(13、14、15)的结构简单,分子较小,在计算机对接模型中未能体现出较好的结合力。总体而言,表2中分值较高的化合物为环十四烷(11)、2-羟基环十五烷酮(12)、β-石竹烯(16)和蛇麻烯(17)。
从受体的角度分析,信号传导与活化转录因子 3(STAT3,PDB ID:1BG1)和白介素-1β 转化酶(Caspase-1,PDB ID:1RWN)与上述化合物的结合力普遍较低,结合能在-3.2~-5.5 kcal/mol之间。而5D3I、4DJH、5C1M、6A93、2GMX等蛋白表现出较强的结合力,结合能在-7.5~-8.3 kcal/mol之间。选取这5个蛋白与化合物11、12、16和17进一步进行折线图分析(见图2)。可以看出,化合物11、16、17对4DJH的结合能力较强,而化合物12对5D3I的结合力最强,为-8.3 kcal/mol。

表2 化合物(1~17)与受体对接打分值(kcal/mol)

图2 化合物11、12、16和17与5种蛋白结合能折线图
3.2 化合物与蛋白对接的示意图
化合物11、16、17与4DJH,12与5D3I对接的三维示意图见图3。图中可见化合物均能较好地卡在空腔中,契合度较高。
在平面图(见图4)中可以发现,化合物11的脂肪环与4DJH中的GLN115、ILE316、TYR320、TYR66、VAL69、THR321、VAL65、ILE62、TYR313等多个氨基酸残基之间存在范德华力;同时脂肪环还有ALA317氨基酸残基形成Alkyl键(图4A)。化合物12的脂肪环与5D3I 中VAL302、ASN267、PHE266、ASP263、PHE325、ASP327、GLY293、ASP294、ASN296、PRO297、THR330等多个氨基酸残基之间存在范德华力;羰基与PHE295形成氢键(图4B)。化合物16与4DJH中的PHE314、SER116、GLN115、TYR66之间存在范德华力;其甲基以及碳环分别与ILE62、TYR119、ALA317形成Alkyl键;碳环双键与TYR119、TYR313氨基酸残基形成Pi-Alkyl键(图4C)。化合物17与4DJH中的PHE314、ILE316、TYR320、SER116、GLN115等氨基酸残基之间存在范德华力;其甲基分别与TYR119、ILE62、ALA317、TYR66等氨基酸残基形成Alkyl键;碳环双键与ALA317和TYR313氨基酸残基形成Pi-Alkyl键;10位CH2与TYR119氨基酸残基形成Pi-Sigma键(图4D)。

注:A:11/4DJH;B:12/5D3I;C:16/4DJH;D:17/4DJH。图3 化合物与蛋白分子对接的三维图

注:A:11/4DJH;B:12/5D3I;C:16/4DJH;D:17/4DJH。图4 化合物与蛋白分子对接的平面图
4 讨论
白芷是我国四十种大宗常用中药材品种之一,在中医临床上常用来治疗各类疼痛,如头痛、面部疼痛和疮疡肿痛方面。多种模型中白芷挥发油均表现优良的抗炎镇痛活性,并且与白芷香豆素联用时有明显的协同作用[5-6]。但不同品种之间、同一品种的不同产地之间、同一产地的不同地块之间的白芷挥发油之间都有明显的差异[16],且多数研究未能将化学组分分析与药理活性测试结合起来。而挥发油的化学组分与所用药材和提取工艺有较大,为了保证挥发油化学组分与药理活性的对应关系,本文对接采用了由同一课题组完成的化学组分数据[17]和药理活性指标[3-4]。白芷挥发油占药材的质量比重较低,因此选取了挥发油中占比0.5%(约占生药材中二十万分之一)[17]以上的成分进行分子对接。
化合物36和41是多种香料的重要成分,在白芷中的含量为0.69%个0.54%,含量虽少,但已有的研究表明,二者具有良好的抗炎等药理活性。其他报道化合物单萜或倍半萜,但含量更低,本文中未进行探讨。化合物75(挥发油中质量百分比1.57%)是化合物45(挥发油中质量百分比4.17%)的衍生物,目前尚未有广泛的药理活性报道。但化合物75结构类似物麝香酮具有显著的抗炎等药理活性[18],提示我们化合物75值得进一步研究其抗炎等活性。白芷挥发油中的倍半萜以及大环类结构是后续研究中重点关注的化合物。
阿片受体(κOR和μOR)是重要的中枢镇痛作用靶点,但尚未发现白芷成分基于κOR或μOR的镇痛研究。本次的分子对接显示,κOR可能与白芷挥发油中的倍半萜等成分具有良好的结合能力,是白芷镇痛的潜在关键结合蛋白。TLR2和JNK-1是抗炎镇痛的作用靶点,本处的分子模拟对接显示化合物36、41、45和75均与TLR2及JNK-1具有良好的结合能力,可能是白芷挥发油抗炎和镇痛的关键蛋白之一。后续的细胞等药理实验中将重点关注这些蛋白。
本文以化合物-靶点相互作用为切入点,揭示了白芷挥发油与抗炎镇痛的特点。即预测了白芷挥发油治疗疼痛的活性成分,又明确了其作用靶点,后续的研究中将采用细胞或动物模型验证上述发现,以期为白芷的研究和开发提供一定的科学依据。
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
中国药学药品知识仓库(2022年7期)2022-05-10
南方医科大学学报(2022年3期)2022-04-13
中国药学药品知识仓库(2022年5期)2022-04-11
亚太传统医药(2022年3期)2022-04-01
中国药学药品知识仓库(2022年2期)2022-03-23
中国药理学通报(2022年3期)2022-03-22
西南农业学报(2021年10期)2021-12-14
保健与生活(2021年9期)2021-08-26
