
金属性二维过渡金属硫化物的溶剂热合成及电催化析氢性能
2021-02-26 13:44:32余强敏张致远罗雨婷成会明刘碧录
高等学校化学学报 2021年2期
余强敏,张致远,罗雨婷,李 洋,成会明,3,刘碧录
(1.清华大学深圳盖姆石墨烯中心,清华⁃伯克利深圳学院/清华大学深圳国际研究生院,深圳518055;2.伍伦贡大学超导和电子材料研究中心,伍伦贡2500,澳大利亚;3.中国科学院金属研究所,沈阳材料科学国家实验室,沈阳110016)
氢能具有能量密度高、产物零碳排放且可再生等优点,被认为是最理想的能量载体之一,开发和利用氢能对能源的可持续发展具有重要意义[1,2].在众多的制氢技术中,电解水制氢优势显著,具有能量转换效率高、氢气(H2)纯度(>99.99%)高及无需苛刻制备条件等优点[3~5].在电解水反应中,电极反应动力学直接关系到整个电解水能量转换效率的高低[6,7].因此,具有高催化活性的电极材料是提高电解水效率的决定性因素.商用贵金属铂基(Pt)催化剂被认为是最理想的析氢反应(HER)电催化材料,因为Pt催化剂的氢吸附自由能(ΔGH*)接近于0 eV,非常有利于反应中间体的吸附和反应产物的脱附.此外,金属Pt催化剂具有良好的导电性,可以实现催化过程中高效的电子转移[8,9].然而,贵金属Pt资源稀缺、价格昂贵,难以满足工业析氢催化材料的大规模使用要求.因此,需要寻求资源丰富、价格低廉、性能优异的材料来替代现有的Pt材料以满足工业化需求.廉价的过渡金属化合物,如金属氧化物(CoOx,FeOx,NiOx)[10,11]、金属磷化物(CoPx,FePx)[12~14]、金属氮化物(MoNx)[15]、金属碳化物(MoxCy)[16]以及金属合金(NixMoy)[17]等,均被用作电解水催化剂,然而,这些材料的本征活性差、活性位点数有限及耐腐蚀性差等缺点严重制约了它们在规模化制氢应用中的潜力.
近年来,二维材料,尤其是二维金属性过渡金属硫化物(TMDCs)材料在电催化析氢应用中逐渐受到关注,具有工业化应用前景[18~20].与其它二维材料相比,金属性TMDCs具有很多独特优势.首先,金属性TMDCs是零带隙材料,导电性好,有利于电化学过程中的电荷传输.其次,金属性TMDCs具有丰富的面内活性位点,密度泛函理论计算表明,一些金属性TMDCs的ΔGH*也与Pt接近[21],是现有材料体系中少有的与Pt性能相当的材料,故而无需其它缺陷或掺杂修饰来增加其催化活性位点的数目[22~25].目前,金属性TMDCs主要是通过化学气相沉积(CVD)法制备,该方法制备的样品质量高,但产量低,因而难以在电催化剂中获得规模化应用.相比于CVD法,液相法合成通常具备规模化制备的优点,是可大量制备金属性TMDCs的潜在重要方法.
本文采用溶剂热法制备不同导电属性的TMDCs催化剂.以金属性NbS2为主要研究对象,研究了反应前驱体的滴加速率对催化剂形貌结构的影响和高温退火处理对催化剂结晶性的影响,考察了其在酸性条件下的电催化析氢活性及稳定性,为金属性TMDCs在高效电解水制氢中的应用提供了新思路.
1 实验部分
1.1 试剂与仪器
五氯化铌(NbCl5,纯度99.5%)、五氯化钼(MoCl5,纯度99.5%)、三氯化钒(VCl3,纯度99.5%)、十二硫醇(DDT,纯度99.0%)和油胺(OLA,纯度85.0%)均购于上海阿拉丁生化科技股份有限公司;硫酸(H2SO4,质量分数98.0%)、丙酮(C3H6O,纯度99.5%)和乙醇(C2H6O,纯度99.8%)均购于国药集团化学试剂有限公司.
SU-8010型场发射扫描电子显微镜(SEM,日本日立公司);Tecnai F30型透射电子显微镜(TEM,美国FEI公司);PHI5000VersaProbeII型X射线光电子能谱仪(XPS,日本Ulvac公司);D8 Advance型X射线衍射仪(XRD,德国布鲁克公司);LabRAM HR型拉曼光谱仪(Raman,激发波长532 nm,日本Horiba公司);VMP-300型电化学工作站(法国Biologic公司).
1.2 实验过程
不同导电属性的二维TMDCs电催化剂通过溶剂热法和高温热处理制备.首先,将25 g油胺和138 mg NbCl5依次加入到100 mL的圆底烧瓶中,向圆底烧瓶中的油胺通入氮气(流量为30 sccm)直至反应结束,使得NbCl5处于氮气环境下避免空气进入,在此过程中通过施加搅拌使NbCl5均匀分散在油胺溶液中.然后将装有油胺溶液的圆底烧瓶加热至280℃恒温后,以不同速度(5.0,1.0,0.2和0.1 mL/min)滴加3.0 mL DDT,继续反应1 h后冷却至室温,加入过量的丙酮洗涤产物,通过离心分离作用洗涤固体样品(重复3次).样品再次用乙醇和去离子水依次清洗并真空干燥烘干.最后将干燥的样品置于管式炉中在不同温度(650,750和850℃)下退火处理2 h,退火升温速率为10℃/min,退火过程中通入氩氢混合气(95 sccm Ar+5 sccm H2);退火处理后冷却至室温取最终产物(NbS2)待用.在相同条件下分别合成出NbS2、二硫化钼(MoS2)和二硫化钒(VS2)3种二维材料.此外,采用相同制备过程将NbS2生长在钼箔(Mo foil)上用作自支撑电极,在样品洗涤过程中,将NbS2/Mo foil浸泡在丙酮和乙醇中12 h后烘干,最后将烘干的样品置于管式炉中,在750℃下通入氩氢混合气(95 sccm Ar+5 sccm H2)退火处理2 h,然后冷却至室温获得最终产物(NbS2/Mo foil).
1.3 电化学析氢性能测试
不同电催化剂的催化性能在室温下采用标准三电极体系进行测试.其中将TMDC催化剂(负载量约为0.3 mg/cm2)负载在玻碳电极上作为工作电极,石墨棒为对电极,饱和银/氯化银为参比电极,0.5 mol/L H2SO4溶液为电解液.向电解液中通30 min氮气(流量为50 sccm),以排除溶液中的其它气体.线性扫描伏安曲线(LSV)的工作电压为0.1~—0.5 V,扫描速率为5 mV/s;电化学阻抗谱(EIS)的电压设为—0.2 V,交流频率范围为0.1~105Hz,振幅为5 mV;循环伏安(CV)曲线以不同扫描速率在相对标准氢电极电位为0.05~0.15 V范围内进行测试;电化学活性面积(ECSA)通过不同扫描速率(10,20,40,60和80 mV/s)下的循环伏安曲线转化得到.采用恒电位法在不同电位下进行长时稳定性(I-t)测试.电化学电位(E)均根据可逆氢电极进行校正(Vvs.RHE),在0.5 mol/L H2SO4溶液中,E(RHE)=E(Ag/AgCl)+0.20 V.
2 结果与讨论
2.1 催化剂的形貌与结构表征
在退火温度为750℃下,不同DDT滴加速率下合成的NbS2如图1所示.由图1(A~D)可见,随着DDT滴加速率的减小,NbS2逐渐由微米颗粒转变为片层结构,最终呈现出二维纳米片组成的“花状”形貌.DDT作为反应还原剂,其滴加速率直接影响到NbS2成核的快慢.当DDT大量滴加(滴加速率快)时,由于反应速率过快,形成的晶核因高的表面能导致其相互聚集而形成块体结构.将DDT滴加速率减慢,成核反应速率也相应变慢,团聚现象逐渐减弱,从而更易形成二维纳米片状晶体结构.采用电化学循环伏安法对以上4种不同DDT滴加速率条件合成的NbS2材料的电化学活性面积进行了测试(图S1,见本文支持信息).图1(E)给出了4种NbS2电催化剂的充放电电流差(ΔJ)与扫描速率的线性关系,根据公式换算后所得的催化剂电化学活性面积如图1(F)所示.当DDT滴加速率为5.0 mL/min时,其电化学活性面积仅为20.2 cm2,随着DDT滴加速率逐渐减至0.1 mL/min时,其电化学活性面积增大至159.5 cm2,展现出二维材料的高比表面积特征,有利于电催化反应过程中的反应前驱体与催化位点充分接触,图1(G)所示为二维NbS2材料及其HER过程示意图.
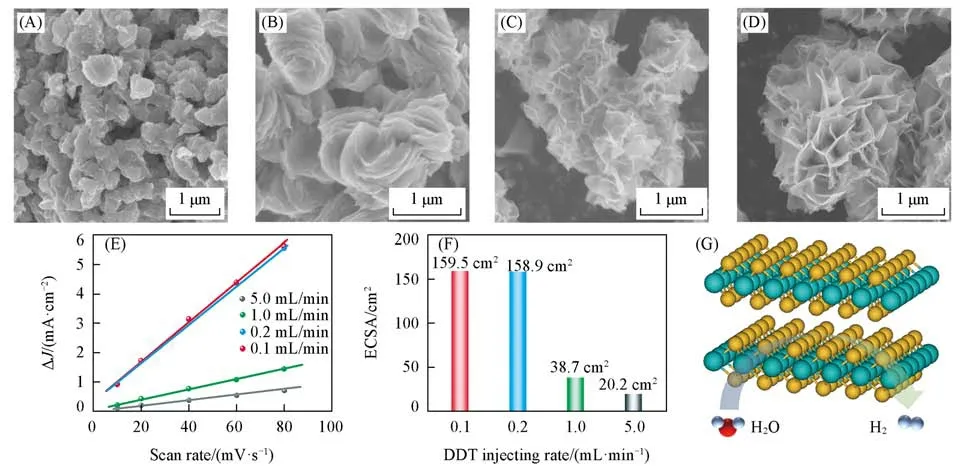
Fig.1 SEM images of two⁃dimensional(2D)NbS2 with different injecting rates of DDT precursors(A—D),capacitive currents at the potential of 0.10 V against the scan rate calculated from Fig.S1(E),ECSA of NbS2 samples with different injecting rates of DDT precursors(F)and schematic of 2D NbS2 for HER(G)
进一步对具有高比表面积的NbS2材料(DDT滴加速率为0.1 mL/min,退火温度为750℃下合成)进行元素分析及晶体结构和原子结构的表征.图S2(见本文支持信息)为NbS2材料的EDS元素成分分布图及相应的谱线,测试结果显示材料成分中S/Nb的原子比为1.93∶1,与理论值2∶1相近.图2(A)为NbS2材料的XRD谱图,4个强衍射峰对应于NbS2的(002),(100),(110)和(200)晶面,与NbS2标准卡片(PDF#41-0980)的衍射峰一致[26,27],表明所制备的NbS2是2H相晶体结构.图2(B)为NbS2材料的Raman光谱图,3个Raman特征峰(E1g,278 cm-1;E2g,330 cm-1;A1g,382 cm-1)与文献[28]报道的2H相NbS2材料一致,与XRD所得的结构相吻合.为进一步确定合成产物的价态,采用XPS测定了NbS2的Nb和S元素的结合能,结果如图S3(见本文支持信息)和图2(C,D)所示.首先,由图S3可见,合成的样品中仅含有Nb,S,C和O 4种元素.图2(C)中Nb3d谱图存在3组峰,分别对应于Nb5+(210.5 eV)、Nb4+(3d3/2,207.7 eV;3d5/2,204.3 eV)以及阴离子插层Nb(4—δ)+(3d3/2,206.5 eV;3d5/2,203.7 eV)[27];图2(D)S2p谱图的2组峰分别对应于S—O(164.7 eV)和S2—(2p1/2,163.3 eV;2p3/2,162.0 eV[29]).以上结果表明,溶剂热合成的主要产物是NbS2材料.对产物进行TEM表征,如图2(E)所示,NbS2呈现二维层状结构;高分辨透射电子显微镜(HRTEM)照片显示NbS2材料具有高质量晶态结构[图2(F)],晶格间距为0.30 nm,对应于2H相NbS2的(100)晶面.此外,对其它2种溶剂热合成的MoS2和VS2材料也进行了HRTEM表征,均呈现出高质量的晶体结构(图S4,见本文支持信息).以上结果表明,溶剂热法及后续高温处理对合成高质量的2H相TMDCs晶体具有普适性.
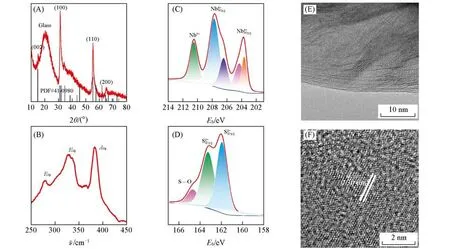
Fig.2 XRD pattern(A),Raman spectrum(B),XPS spectra of Nb3d(C)and S2p(D),TEM(E)and HRTEM(F)images of metallic 2D NbS2 materials
2.2 电化学析氢性能
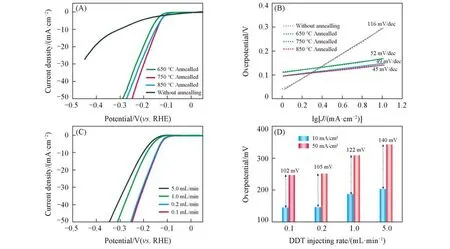
Fig.3 LSV curves(A)and corresponding Tafel curves(B)of NbS2 with different annealing temperatures,LSV curves(C)of NbS2 with different injecting rates of DDT precursors and overpotentials(D)of NbS2 at 10 and 50 mA/cm2 under different injecting rates of DDT precursors
为评价不同条件下合成的二维NbS2材料的电催化析氢性能,测试了其催化活性和动力学特性,结果见图3.图3(A)为DDT滴加速率为0.1 mL/min时不同退火温度处理的NbS2的线性扫描伏安曲线.与未经退火处理的材料相比,退火处理后的NbS2材料的催化活性大幅提高;在退火处理温度为750℃时性能达到最佳,该样品在电流密度为10 mA/cm2时过电位仅为146 mV,远小于同等条件下未经退火处理的样品(在10 mA/cm2时过电位为305 mV).当退火温度高于或低于750℃时,样品的催化活性均减弱.进一步评价了不同退火温度处理的NbS2电催化剂在HER中的动力学特性,如图3(B)所示.根据塔菲尔方程η=a+blgJ[其中,a(mV)为电流密度为单位数值(1 mA/cm2)时的过电位值;J(mA/cm2)为电流密度;b(mV/dec)为塔菲尔常数,b值越小,催化剂活性越高]计算了其在HER过程的塔菲尔斜率.在电催化析氢过程中,750℃退火处理后的NbS2催化剂的塔菲尔斜率为45 mV/dec,远小于未经退火处理的NbS2材料的116 mV/dec,表明750℃退火处理的NbS2表现出优异的动力学特性.此外,对不同退火温度处理的NbS2电催化剂的电荷传输能力进行评价,结果如图S5(见本文支持信息)所示,可见,750℃退火条件下NbS2的电荷传输电阻约为30Ω,小于850℃的35Ω,远小于650℃的63Ω和未退火样品的106Ω,表明750℃退火条件下的NbS2具有更快的电子转移速率.根据以上结果可知,高温退火处理可提升材料的导电性和结晶度,提高材料的催化活性.相反,过高温度的还原气氛处理不利于保持材料原始的本征结构,如图S6(见本文支持信息)所示,与750℃下退火后的NbS2样品相比,850℃退火后的样品其Nb3d峰整体向低结合能方向偏移约1.0 eV,表明NbS2中的Nb从本征的+4价态转变为更低价态,从而改变了NbS2的本征结构;另外,金属性NbS2本征结构的改变使得NbS2的导电性降低,进而使其催化活性降低.除了退火条件,对不同DDT滴加速率下制备的NbS2的析氢催化活性进行了电催化性能表征,其具体变化趋势如图3(C)所示.随着DDT滴加速率的减小,NbS2的催化活性逐渐提高,原因在于:一方面滴加速率慢有利于二维薄层结构的形成,从而暴露更多催化活性位点;另一方面,“花状”多孔结构的形成有利于反应中质子的传输及气体扩散,可进一步提高HER速率.此外,由图3(D)可见,当电流密度从10 mA/cm2增加到50 mA/cm2时,“花状”多孔结构NbS2所需电位只增加了102 mV,远小于微米颗粒NbS2的140 mV,进一步表明“花状”多孔结构NbS2具有更快的HER反应动力学.

Fig.4 LSV curves(A)and corresponding Tafel curves(B)of MoS2,VS2,NbS2 and Pt/C catalysts,EIS curves of MoS2,VS2 and NbS2 catalysts(C)and the stability curve of NbS2 catalysts at the potential of-0.146 V(vs.RHE)(D)
基于以上研究结果,通过优化实验参数制备了不同种类的TMDCs材料,包括金属性的二维NbS2和VS2,以及半导体性的二维MoS2.对优化后的3种不同TMDCs进行了HER电催化活性表征.如图4(A)所示,在电流密度10 mA/cm2下,NbS2、VS2和MoS2的过电位分别为145,148和201 mV,表明金属性TMDCs具有更高的反应活性,与文献报道一致[21];图4(B)为NbS2、VS2和MoS23种催化剂的塔菲尔曲线,其斜率分别为45,50和63 mV/dec,说明3种不同TMDCs催化剂的HER过程均遵循Volmer-Heyrovsky的反应机理[30].图4(C)为3种TMDCs催化剂的Nyquist图,图中半圆直径表示电极电荷转过移电阻Rct,插图为实际测试电化学阻抗的拟合模型.测试结果显示,NbS2和VS2的Rct分别为30和32Ω,远低于MoS2的64Ω,表明金属性TMDCs催化剂具有良好的电子传输能力和HER动力学.除电催化活性外,电极材料的稳定性和耐久性也是评价电催化剂实际应用价值的重要依据.图4(D)为NbS2催化剂的稳定性和耐久性测试曲线,在恒电压下连续工作24 h后,NbS2催化剂的电流密度几乎未衰减.此外,在循环5000次后,NbS2催化剂在10 mA/cm2电流密度下所需过电位基本没有增大.对电化学循环后的NbS2催化剂进行形貌结构表征,如图S7(见本文支持信息)所示,样品在经过电化学循环后无明显团聚现象,其晶格结构无明显变化,该结果表明NbS2催化剂具有良好的电化学稳定性.以上结果均表明,相较于半导体性TMDCs,金属性TMDCs在HER中具有更高的催化活性和应用前景.
为进一步验证溶剂热法制备的金属性TMDCs在实际析氢反应中的应用前景,将生长在钼箔基底上的NbS2(NbS2/Mo foil)自支撑电极置于高电流密度析氢条件下进行测试,结果如图5(A)所示.在400 mA/cm2的高电流密度下,NbS2/Mo foil所需过电位仅为386 mV,略高于同等条件下Pt催化剂的303 mV.为验证制备的NbS2/Mo foil自支撑电极的稳定性,对其进行了长时间计时电流测试.如图5(B)所示,在过电位为225 mV下持续工作24 h,NbS2/Mo foil的电流衰减率仅为0.1 mA/h,该结果证明NbS2/Mo foil自支撑电极具有良好的电化学稳定性.催化材料的规模化制备是衡量其工业应用中的重要指标[31,32],基于此,不同催化剂制备方法的产能需进一步评价.目前制备金属性TMDCs的方法包括CVD法、溶剂热法和化学气相输运(CVT)法.不同方法制备的金属性TMDCs展现出不同的析氢活性,如图6和表1所示,在电流密度为10 mA/cm2时,CVD法和溶剂热法制备的金属性TMDCs在电流密度为10 mA/cm2下的析氢过电位均小于200 mV,优于同等条件下CVT法制备样品的过电位(300~1000 mV).从表1可见,CVT和溶剂热法制备的金属性TMDCs可实现10~100 mg/h的生产速率,高出CVD法2~3个数量级.
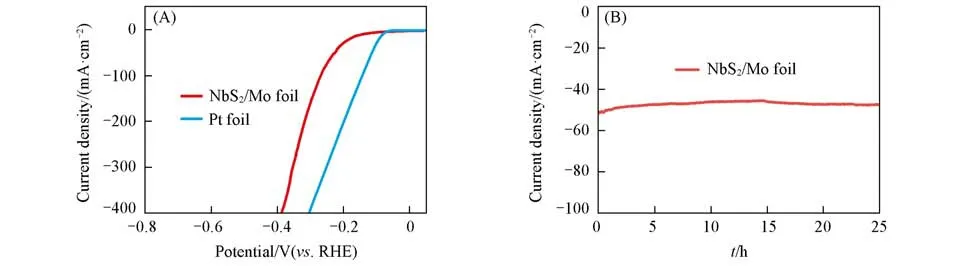
Fig.5 LSV curves(A)and I⁃t curve(B)of self⁃supporting electrode of NbS2/Mo foil
综上,溶剂热法是规模化制备高性能金属性TMDCs电催化剂的最佳方法,具有潜在的工业化应用前景.
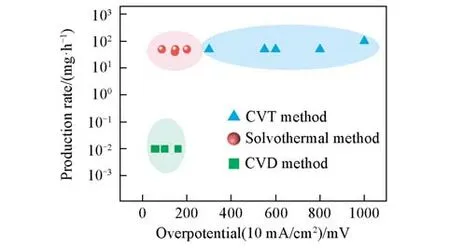
Fig.6 Performance and production rate of different TMDCs by CVT,CVD and solvother⁃mal methods
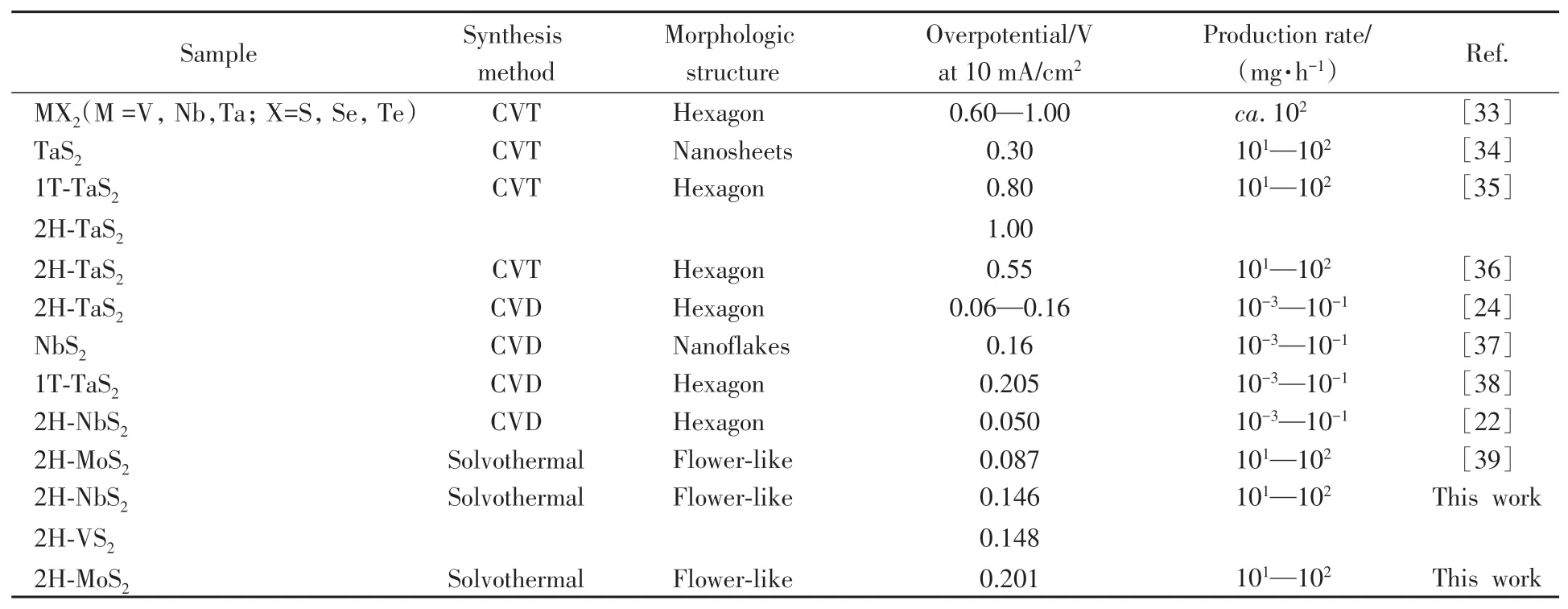
Table 1 Summary of synthesis methods,sample description and overpotentials of TMDCs for HER
3 结 论
采用溶剂热法制备出不同导电属性的TMDCs析氢电催化剂.通过调控DDT反应前驱体的滴加速率和产物高温退火处理,可显著提高NbS2的催化性能.酸性电解质中,当电流密度为10 mA/cm2时,NbS2所需的析氢过电位仅为146 mV;动力学分析发现,该类催化剂遵循Volmer-Heyrovsky反应机理.通过对比TMDCs材料的3种主要合成方法,发现溶剂热法制备的TMDCs与CVD法制备的材料催化性能相当,并远优于CVT法制备的TMDCs材料.值得指出的是,溶剂热法制备TMDC的产率比CVD法高2~3个数量级.表明溶剂热法制备的TMDC电催化剂在实际电解水析氢技术中具有良好的应用前景.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200454.
猜你喜欢
中国民间疗法(2021年6期)2021-06-09 06:18:58
新课程·下旬(2017年11期)2018-01-22 19:30:55
新课程(下)(2016年5期)2016-08-15 02:57:43
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
应用化工(2014年12期)2014-08-16 13:10:46
无机化学学报(2014年6期)2014-02-28 17:31:57
无机化学学报(2014年4期)2014-02-28 17:31:23
无机化学学报(2014年3期)2014-02-28 17:30:46
应用技术学报(2014年1期)2014-02-28 14:52:11
