
炎性肠病易感基因GPR35在肠炎发生发展中的功能研究
2021-02-26 00:00郑燕森卓林刚李大力刘明耀
遗传 2021年2期
郑燕森,卓林刚,李大力,刘明耀
研究报告
炎性肠病易感基因在肠炎发生发展中的功能研究
郑燕森,卓林刚,李大力,刘明耀
华东师范大学生命科学学院生命医学研究所,上海调控生物学重点实验室,上海 200241
炎性肠病在全球范围内发生极其普遍,具有反复发作、难以治愈的特点,也是诱发结直肠癌的高风险因素之一。肠炎的发生与遗传因素密切相关,有报道发现位于基因座上的多个单核苷酸多态性(single nucleotide polymorphism, SNP)位点rs4676410、rs3749171和rs3749172与肠炎敏感性高度相关,但是基因在肠炎的发生发展进程中的功能及相关机制尚没有明确结论。为了研究在肠炎中的作用,首先通过CRISPR/Cas9技术构建敲除小鼠,随后利用DSS诱导的肠炎模型评价在肠炎发生中的作用,发现敲除小鼠在体重变化、DAI评分、肠上皮损伤以及炎性细胞浸润等肠炎相关指标显著低于野生型小鼠。为了研究肠炎相关SNP突变对GPR35活性的影响,首先根据rs3749171和rs3749172SNP位点突变信息构建GPR35-T108M和GPR35-S294R两种突变型受体,其次通过多种下游信号通路活性测试,发现两种突变均能够增强GPR35受体活性。最后通过Western blotting分析发现相较于野生型小鼠,敲除小鼠肠上皮Erk1/2磷酸化水平增加,表明敲除后可能通过上调Erk1/2信号通路的方式抑制肠炎的发生发展。综上所述,本研究发现人类肠炎易感的rs3749171和rs3749172位点可能通过激活GPR35及下游信号通路的方式促进肠炎的发生发展,为炎性肠病的治疗提供了潜在的药物作用靶点。
rs3749171;rs3749172;;肠炎;动物模型
炎性肠病(inflammatory bowel disease, IBD)是一种全世界范围内高发的肠道系统炎症,分为克罗恩病(Crohn disease, CD)和溃疡性结肠炎(ulcerative colitis, UC)两种疾病亚型,可导致腹泻、直肠出血、腹痛和营养不良等症状[1]。此外,IBD是结直肠癌(colorectal cancer, CRC)的主要危险因素,IBD患者的结肠癌发病率是普通人群的18倍[2~4]。鉴于全世界范围内IBD的发病率不断增加并带来巨额的医疗负担,肠炎已成为目前亟需解决的公共健康问题。因此,开展肠炎发病机制的研究及开发IBD治疗药物靶点对肠炎的预防及治疗方面有着极其重要的意义。
目前研究已发现,肠炎发病主要是由于吸烟、肥胖、饮食习惯等外界环境因素及肠炎易感基因和等遗传因素导致肠内微生物稳态破坏,肠上皮屏障功能缺失以及先天/适应性免疫系统功能紊乱等功能障碍,最终导致肠炎的发生发展[5~7]。鉴于基因突变在肠炎发生发展进程中发挥了重要作用,全基因组关联研究(genome-wide association studies, GWAS)已被应用于肠炎发病风险预测及机制研究中[8]。通过对与炎性肠病相关的单核苷酸多态性(single nucleotide polymorphism, SNP)位点调研,本课题组前期发现3个独立的报导均表明位于基因座的SNP位点与肠炎高度相关,但是并未研究清楚是否发生活性突变及表达水平变化[9~11]。在dbSNP数据库分析上述SNP位点可以发现:rs4676410和rs3749172位点在亚洲群体出现频率较高,GWAS统计出现频率分别为30.6% (样本量124)及74.1% (样本量324),而对应的欧美群体中为19.5% (样本量97104)及54.6% (样本量123608);而rs3749171位点在欧美群体中GWAS统计出现频率16.8% (样本量95590)高于亚洲群体相关位点出现频率4.3% (样本量140)[12]。其中rs4676410位于内含子区域,可能通过影响转录调控等方式影响目的基因的表达[9];而rs3749171和rs3749172位于编码区,导致GPR35发生T305M和S294R位点氨基酸突变,可能会对GPR35受体活性产生影响[10,11]。
GPCR家族作为最大的一类细胞膜受体家族,能够对光感应、信息素、激素、神经递质在内的多种刺激产生反应并调控细胞增殖、凋亡、迁移以及应激反应,与多种疾病发生发展相关[13]。是G蛋白偶联受体(G protein coupled receptor, GPCR),属于G蛋白偶联受体家族视紫红质样受体亚群[14]。在外周血白细胞、脾脏、小肠、结肠和胃等多个组织器官及细胞类群中表达较高,且被证明可能参与哮喘、心血管疾病、肠炎、糖尿病等一系列疾病的发生发展,但具体功能及机制尚未研究清楚[14]。目前为止,在肠炎的病理进程中的作用及机制依旧没有确定的结论。GPR35的激动剂包括犬尿酸,LPA,Zaprinast以及双羟萘酸,但是不同激动剂在肠炎发生发展进程中的作用完全相反:有研究证明社交压力带来的犬尿酸积累能够通过GPR35促进肠炎的发生发展,而双羟萘酸可以通过促进肠上皮粘膜修复的方式抑制肠炎的发生发展[15,16]。鉴于激动剂相关的研究无法证明在肠炎发生发展中的作用,构建基因突变小鼠,并在敲除动物模型水平上进行肠炎模型构建将有助于明确在肠炎发生发展中的功能,鉴定新的药物作用靶标。
在肠炎的发病进程中,GPCR下游信号通路如ERK1/2、RHOA及AKT等信号通路等在肠炎发生发展中能够起到重要的作用[17]。ERK1/2信号通路在肠上皮组织的激活能够促进肠上皮粘膜修复,进而保护肠道组织免受肠炎症状侵袭[18]。有研究显示GPR35能够通过下游Gq、Gi/o、G12/13以及β-arrestin分子调控下游ERK1/2和 AKT等信号通路活性。在DSS诱导肠炎模型中,是否能够通过调控ERK1/2信号通路活性调节肠炎发病进程成为需要研究的重点。本研究将从细胞及动物模型两个方面探究肠炎相关SNP位点对活性水平的调控及在肠炎发生发展中的作用及相关机制,为肠炎易感基因的鉴定、肠炎发生发展机制的探索以及后续肠炎治疗药物靶点开发打下理论基础。
1 材料与方法
1.1 载体构建
人源有两个转录异构体,根据文献查阅及序列比对,选择亚型作为后续研究蛋白,cDNA全长930 bp,蛋白全长309 aa。根据人源cDNA序列设计反转录引物及突变引物,引物交由上海生工生物工程技术服务有限公司合成。利用点突变试剂盒的方法构建GPR35 T108M,S294R突变载体,最后将GPR35 WT,T108M及S294R序列利用常规分子生物学技术克隆至pcDNA3.1载体(V79020,Thermo Fisher,美国)中,用于后续活性研究。引物序列见表1。

表1 GPR35野生型及突变型克隆引物序列
1.2 细胞培养及转染
293T细胞购买自上海中科院。将状态良好的293T细胞接种到含10%胎牛血清,l%青霉素/链霉素的DMEM培养基。将细胞放置于37℃,5% CO2的培养箱中培养。细胞密度到达90%后,利用1:3的比例传代培养。待细胞密度达到60%~70%后,利用转染试剂PEI将质粒转染细胞到细胞内。其中,质粒与PEI按照1∶3的比例混合加入细胞培养基中,8 h后换新鲜培养基。24~48 h后转染质粒蛋白表达水平达到高峰且效果持续到72~96 h。
1.3 β-arrestin招募检测
本实验主要参照Mitsuru Hattoriw论文中的方法检测野生型及突变型GPR35受体招募β-arrestin能力[19]。将GPR35、GPR35T08M、GPR35S294R序列克隆至ELuC载体中。293T细胞密度达到60%后,利用PEI将GPR35-ELucC和ELucC-ARRB2转染到细胞中。24 h后将细胞接种于96孔板中,待细胞贴壁后进行β-arrestin招募实验。将不同浓度Zaprinast及荧光素酶底物加入培养基中,利用酶标仪检测细胞生物荧光强度。
1.4 钙流检测实验
将GPR35,GPR35T08M,GPR35S294R质粒与Gq质粒转染进293T细胞中,24 h后将细胞接种到96孔板中,每孔2×104个细胞。转染48 h后,加入不同浓度的Zaprinast,利用Fluo-8钙流检测试剂盒检测各组细胞中钙流活性。
1.5 CRE荧光素酶报告系统检测
将GPR35、GPR35T08M、GPR35S294R质粒及CRE-Luc质粒转染进293T细胞中,24 h后将细胞接种于96孔板,每孔2×104个细胞。待细胞贴壁后,加入不同浓度Zaprinast至培养基中,24 h后根据Dual Luciferase Reporter Assay System操作说明书,利用酶标仪检测各组细胞荧光强度。
1.6 Gpr35敲除小鼠构建
根据CRISPR/Cas9系统sgRNA设计原则,针对鼠源基因第6个外显子部分通过Zhang lab (http://crispr.mit.edu)网站设计sgRNA靶点。sgRNA序列为sgRNA1:GGGCCTGCTGCTCAACAGCGT GG, sgRNA2:CTATATGACCAACCTGGCTGTGG。sgRNA引物交由上海生工生物工程技术服务有限公司合成。利用显微注射技术将sgRNA及Cas9蛋白注射进受精卵中,并将存活的受精卵移植到假孕受体母鼠内,至母鼠生产得到F0代。提取小鼠组织DNA进行PCR鉴定,检测Cas9敲除效果并繁殖获得敲除小鼠。
1.7 DSS小鼠肠炎模型构建
利用3%硫酸葡聚糖(dextran sulfate sodium, DSS)饲喂6~8周同窝小鼠5天。每日称重小鼠体重,观察小鼠粪便粘稠程度及便血情况。5天后将3%DSS水替换成正常的饮用水。第7天利用CO2窒息法处死小鼠,解剖并测量小鼠结肠长度,收集小鼠结肠组织用于后续实验。
1.8 实时荧光定量PCR分析
依照Trizol试剂盒说明书提取小鼠结肠组织总RNA,分光光度仪检测RNA浓度。利用TAKARA反转录试剂盒将定量RNA反转为cDNA。依照TaKaRa实时荧光定量PCR (Quantitative Real-time PCR, qPCR)检测各类炎症因子表达水平。qPCR引物见表2。
1.9 HE染色
新鲜小鼠结肠组织利用5%多聚甲醛固定,梯度酒精脱水,二甲苯,石蜡包埋。样品蜡块组利用切片机切片,厚度为5 μm。利用苏木精/伊红对石蜡切片进行染色,染色结果采用显微镜进行观察拍照。

表2 qPCR引物
1.10 免疫组化及免疫荧光染色
利用S-P法对小鼠结肠石蜡切片进行免疫组化及免疫荧光染色。应用BrdU抗体(ab6326,abcam,美国),Lysosome抗体(ab24170,Abcam,美国),稀释比例分别为1∶500,1∶300。随后利用DAB显色,苏木精复染细胞核,普通光学显微镜下观察并记录染色结果。免疫荧光实验中F4/80抗体(70076T,CST,美国)稀释浓度为1∶200,二抗为绿色荧光标记二抗,DAPI复染细胞核,荧光显微镜下观察染色结果。
1.11 Western blotting (WB)分析
结肠组织利用液氮研磨粉碎组织,利用RIPA加蛋白酶磷酸酶抑制剂(100×)裂解组织蛋白。BCA蛋白定量试剂盒进行蛋白定量。将等量蛋白样品上样,PAGE胶分离蛋白,湿转法将蛋白转到NC膜中。ERK1/2 (9102S,CST,美国),p-ERK1/2 (8544S,CST,美国)抗体浓度为1∶1000,β-actin (3700T,CST,美国)抗体浓度为1∶5000。荧光标记羊抗兔二抗浓度为1∶5000。利用Odyssey成像系统分析结果。
1.12 数据处理
2 结果与分析
2.1 GPR35相关肠炎SNP位点将导致GPR35编码蛋白突变或表达水平变化
针对基因座上肠炎相关的SNP位点进行位置分析发现:rs4676410位点位于第5个内含子的位置,而rs3749171和rs3749172位点位于的编码区(图1 A)。对这3个SNP位点进行分析可以发现:rs46776410位点在基因内含子区域,在UCSC Genome Browser网站查询这个位点时发现rs4676410位点位置存在一个EH38E2090026远端增强子结构序列。rs4676410位点有可能导致表达水平变化[8]。rs3749171和rs3749172 SNP位点位于编码区上,根据对序列比对发现这两个SNP位点会造成GPR35蛋白在108位置的苏氨酸突变为甲硫氨酸(T108M),294位置的丝氨酸突变为精氨酸(S294R)[10,11]。这个突变可能会导致GPR35蛋白结构上的改变,影响GPR35蛋白的受体活性进而调控肠炎的发生发展(图1 B)。因此,基于肠炎相关SNP位点在基因座位置的分析可以推测GPR35活性或表达水平变化是导致肠炎的发生发展的关键原因。
2.2 Gpr35敲除小鼠构建
为了进一步探究在肠炎发生发展中的作用,利用CRISPR/Cas9技术构建了敲除小鼠模型。如图所示,在第6个外显子的编码区域选择两个靶点,并设计相应sgRNA (图2 A)。将体外转录的sgRNA与Cas9蛋白通过显微注射的方法注入受精卵,并将受精卵移植到假孕小鼠体内。最终,在F0代中得到了5只成功敲除的小鼠.通过测序分析发现这5只小鼠体内包含10种不同的突变形式(图2 B)。后续在F1代中寻找能够稳定遗传的突变个体,选择3号小鼠(缺失104 bp)后代相互交配,最终获得纯合突变小鼠开展后续研究(图2C)。
2.3 Gpr35敲除不会引起自发肠炎
成功构建敲除小鼠后,首先对敲除小鼠进行生理观察,结果发现小鼠外形、生长速度、体长等没有明显缺陷。接下来对小鼠小肠,结肠切片染色观察发现:敲除小鼠在肠上皮完整性方面与野生型小鼠并无差异(图3 A)。小肠上皮隐窝中潘氏细胞(lysosome免疫组化标记)平均数目(WT组平均4.5±1.0个;–/–组平均4.6±1.2个,=0.804),绒毛上杯状细胞(Alcian blue染色标记)平均数目(WT平均16.2±2.6个,–/–组平均16.6± 2.3个,=0.7845)及结肠上皮每个视野内杯状细胞(Alcian blue染色标记)(WT平均68.6±11.25个,–/–组平均66.4±13.6个,=0.8234),及位置与野生型小鼠无明显差异(图3 B,C)。这说明敲除后在正常状态下不会造成肠上皮完整性破坏及自发肠炎的发生。

图1 肠炎相关SNP位点分析
A:肠炎相关SNP位点位置分析;B:rs3749171,rs3749172位点造成GPR35氨基酸突变位置分析。

图2 CRISPR/Cas9系统构建Gpr35敲除小鼠
A:构建敲除小鼠的sgRNA靶点序列。下划线表示sgRNA序列,蓝色字体表示PAM序列;B:F0代小鼠靶点附近的基因组测序分析结果;C:3号F0代小鼠(104 bp缺失)与野生型小鼠产生的F1代杂合子小鼠相互交配后F2代小鼠的基因型鉴定,野生型条带为396 bp,敲除条带为282 bp,WT为野生型,HZ为杂合子,HO为纯合子。
2.4 Gpr35敲除能够减缓DSS诱导肠炎模型发生发展
随后,利用DSS诱导的肠炎模型研究在肠道完整性被破坏并发炎症的状况下是否参与到肠炎发生发展的进程中。利用3%DSS替代饮用水,5天后替换为纯净水构建小鼠DSS肠炎模型,两组小鼠在第8天时CO2窒息处死小鼠,解剖并测量两组小鼠结肠长度,取部分结肠组织用于HE染色,免疫组化染色及免疫荧光分析。在建模期间观察记录敲除小鼠及野生型小鼠体重变化,粪便硬度及便血情况。
结果发现,在DSS诱导下敲除组的小鼠在第8天时平体重下降水平明显低于野生型组小鼠(WT组平均体重比重78.6±6.0%,Gpr35组平均比重91.1±4.4%,<0.001) (图4 A);与此相对应的,小鼠日常活动指数(disease activity index,DAI)也显示出Gpr35小鼠肠炎严重程度弱于野生型小鼠(WT组DAI分数6.3±1.1,–/–组DAI分数3.2± 1.3,<0.001) (图4 B);在DSS诱导的肠炎模型中,结肠缩短程度也是评价肠炎表型的方法,对两组小鼠结肠长度进行测量可以发现野生型小鼠组结肠长度远低于敲除小鼠组(WT组平均长度4.9± 0.5 cm,–/–组平均5.7±0.3 cm,<0.01) (图4 C)。对两组小鼠结肠部位切片分析可以发现:野生型小鼠组中结肠组织肠上皮结构破坏程度,总体溃疡面积以及炎症细胞浸润水平远高于敲除小鼠组 (图4 D)。而相应的组织学评分也证明敲除小鼠组肠炎表型更轻微(WT组组织评分分数6.6±1.3,–/–组组织评分4.0±0.8,<0.01) (图4 E)。
随后利用qPCR检测两组小鼠结肠炎症因子表达水平,结果显示敲除小鼠组结肠组织肠炎相关炎症因子如(WT组1.0±0.14,Gpr35组0.3±0.15,<0.001),(WT组1.0±0.13,Gpr35组0.25±0.1,<0.001)(WT组1.0±0.19,Gpr35组0.4±0.08,<0.001)以及(WT组1.0±0.12,Gpr35组0.67±0.29,<0.05)表达水平低于野生型小鼠组,同时的表达量高于野生型小鼠(WT组1.0±0.23,Gpr35组3.12±0.77,<0.001) (图5 A)。对结肠组织进行F4/80免疫荧光结果显示,敲除组小鼠结肠粘膜巨噬细胞浸润数目显著低于野生型组小鼠(WT组平均每个视野52.5±7.2个巨噬细胞,–/–组平均每个视野22±3.2个巨噬细胞,<0.001) (图5 B)。相应的,BrdU免疫组化染色也证明在敲除小鼠在DSS诱导的肠炎进程中肠上皮细胞也保持着较高的增殖水平(WT组平均每个视野87±11.6个BrdU阳性细胞,–/–组平均每个视野141±15.4个阳性细胞,<0.001) (图5 C)。上述结果均证明敲除后能够显著抑制DSS诱导的肠炎模型发生发展。

图3 自然状态下Gpr35敲除小鼠与野生型小鼠小肠,结肠上皮完整性及功能细胞染色分析
A:小鼠小肠,结肠HE染色;B:小肠lysosome免疫组化染色(截图部分为隐窝区域放大图)及小肠,结肠Alcian blue染色,标尺长度200 μm;C:各组细胞染色计数统计分析。其中,WT为野生型对照组小鼠,Gpr35为敲除小鼠组;为无显著水平差异。

图4 DSS诱导肠炎模型体重变化,DAI指数,结肠长度及HE染色分析
利用3%DSS水溶液饲喂两组小鼠,第5天换成正常饮用水,第8天处死小鼠取材。实验期间观察小鼠体重变化等各项指标。A:两组小鼠每日体重变化(以第1天体重为基准);B:两组小鼠每日DAI活动指数评分;C:第8天实验中止后小鼠结肠长度测量;D:HE检测两组小鼠肠上皮结构,标尺长度2000 μm;E:组织学评分评价两组小鼠肠上皮损伤水平。其中,WT为野生型对照组小鼠,Gpr35为敲除小鼠组;*:<0.05, **:<0.01, ***:<0.001。
2.6 Gpr35敲除能够激活ERK1/2信号通路通路抑制肠炎的发生发展
ERK1/2信号通路是GPCR下游重要的信号通路组成部分,GPCR能够通过Gi/o,Gs及β-arrestin 等第二信使因子调控ERK1/2信号通路[20]。WB检测两组小鼠结肠组织中Erk1/2及p-Erk1/2表达水平发现:相较于野生型小鼠组,敲除后能够显著促进结肠上皮细胞Erk1/2磷酸化水平(图6)。这说明敲除后能够通过上调下游Erk1/2相关信号通路活性的方式抑制肠炎的发生发展。
2.7 rs3749171和rs3749172位点突变增强GPR35受体活性
为了探究上述两个SNP突变对GPR35受体活性的影响,构建了GPR35,GPR35T108M及GPR35S294R表达载体。利用Zaprinast作为GPR35受体激动剂,分别采用钙流检测(GPR35下游Gq相关信号通路),β-arrestin招募(GPR35下游β-arrestin招募相关信号通路)以及CRE荧光素酶报告系统(cAMP响应元件、GPR35下游Gi/o和Gs共同调节的信号通路) 3种方式研究突变对GPR35受体活性的影响。结果表明:GPR35T108M,GPR35S294R突变体的受体活性在不同浓度Zaprinast刺激下,下游信号通路活性如钙流响应强度(图7 A),β-arrestin招募水平(图7 B)以及下游CRE表达抑制(图7 C)方面均强于野生型GPR35 (表3)。这些结果提示rs3749171与rs3749172SNP位点突变能够增强GPR35对不同浓度Zaprinast激动剂敏感性及下游信号通路活性。

图5 DSS诱导肠炎模型中炎症因子表达水平,巨噬细胞浸润水平及细胞增殖分析
A:qPCR分析两组小鼠结肠组织炎症因子表达水平;B:免疫荧光染色分析巨噬细胞在两组小鼠肠上皮粘膜中浸润水平,标尺长度100 μm;C:免疫组化分析两组小鼠肠上皮增殖水平,标尺长度200 μm。其中,WT为野生型对照组小鼠,Gpr35为敲除小鼠组;*:<0.05, ***:<0.001。
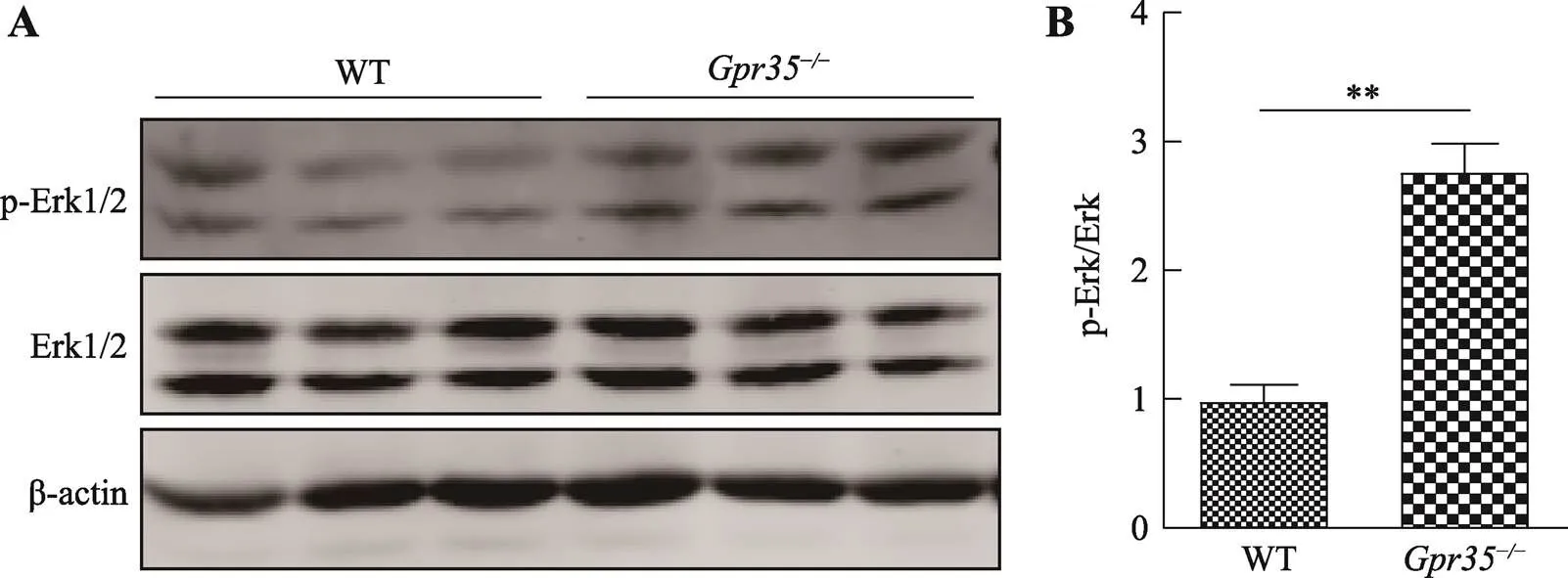
图6 Gpr35敲除对DSS诱导肠炎模型中Erk1/2信号通路活性影响
A:WB分析两组小鼠结肠Erk1/2及p-Erk1/2蛋白表达水平;B:灰度分析两组小鼠p-Erk1/2表达量占总体Erk1/2蛋白比例。其中,WT为野生型对照组小鼠,Gpr35为敲除小鼠组;**:<0.01。

图7 rs3749171与rs3749172突变位点对GPR35受体活性影响分析
A:钙流检测GPR35、GPR35T108M和GPR35S294R在不同浓度Zaprinast刺激下钙流响应强度;B:β-arrestin招募实验检测GPR35、GPR35T108M和GPR35S294R在不同浓度Zaprinast刺激下荧光强度;C:CRE荧光素酶报告系统检测GPR35、GPR35T108M和GPR35S294R在不同浓度Zaprinast刺激下CRE转录活性。为无显著性差异,*:<0.05, ***:<0.001。

表3 GPR35、GPR35T108M及GPR35S294R受体活性变化(归一化处理)统计及显著性分析
value指GPR35T108M,GPR35S294R与GPR35受体活性对比显著性分析结果。
3 讨论
肠炎是一种受多种机制调控的慢性炎症疾病。目前肠炎的治疗手段主要包括糖皮质激素,炎症因子抗体,以及菌群调节等方法。上述治疗方法都能够在一定程度上缓解肠炎表型,但是依旧存在治疗价格高,见效慢,效果差等缺点[21~23]。因此,研究肠炎发生发展机制及治疗靶点筛选依旧是一个很重要的课题。随着基因组分析及测序技术的发展,越来越多的研究开始采用GWAS技术研究肠炎易感的SNP位点,并将其应用于肠炎风险预测及后续机制研究[8]。目前已经筛选出上百个肠炎相关SNP位点,但是这些SNP位点调控基因表达,活性以及后续影响肠炎的发生发展的机制依旧需要大量的工作去验证分析。
GPCR家族作为最大的一类细胞膜受体家族,能够调控多种细胞生理进程[13]。30%的现代药物以GPCR作为治疗靶点,而作为GPCR家族成员的一员,具有巨大的研究意义及药物开发前景[24]。在肠炎方面,GWAS研究分析证明基因座上多个SNP位点与肠炎发生高度相关,但是对于在肠炎发生发展进程中的作用一直没有一个统一的认知[9,10,11,15]。本研究通过CRISPR/Cas9技术构建敲除小鼠用于研究在肠炎发生发展中的作用。结果发现:正常情况下Gpr35小鼠肠上皮完整性及细胞构成与野生型小鼠没有差异,不会引起自发肠炎。但是在DSS肠炎诱导模型中,敲除小鼠在小鼠体重下降水平,DAI评分,结肠长度缩短程度,炎症因子表达水平以及巨噬细胞浸润等多个炎症损伤评价标准远低于野生型小鼠,这说明能够促进肠炎的发生发展。而这个结果与社交压力积累犬尿酸通过促进肠炎发生发展的研究中促进肠炎发生发展的研究结果一致[15]。而对于双羟萘酸的矛盾性结果,有研究证明双羟萘酸是人源GPR35特异激动剂,而对鼠源Gpr35基本没有激动效果[25]。这个可以解释双羟萘酸在DSS小鼠肠炎模型中起到的保护作用这个矛盾的结果。因此,结合本文研究与文献分析结果,最终可以确认在肠炎的发生发展的进程中起到促进作用。
本研究对肠炎SNP易感位点进行筛选,发现3个肠炎相关SNP位点位于基因范围内,其中rs3749171和rs3749172位于编码区,最终导致GPR35突变。针对rs3749171位点,研究人员比对了不同种属在该位置的氨基酸保守性,发现GPR35在108位置的苏氨酸位点在不同种属不具备进化保守性,这预示着GPR35T108M突变可能会导致GPR35活性的改变[10]。该突变位置在GPR35第3个跨膜区位置,该位点发生突变可能会导致GPR35受体识别部位空间位阻发生改变,导致GPR35对配体敏感性发生变化。对于rs3749172位点,研究人员也证明该位点突变与动脉粥样硬化及冠状动脉疾病风险具有相关性,而对其突变氨基酸位置进行分析则发现其突变位置位于GPR35的C末端,294位的丝氨酸突变为酪氨酸可能与GPR35与下游G蛋白及β-arrestin结合活性产生影响最终导致GPR35受体活性改变[26]。
GPR35与炎症及肠炎发生发展的相关机制目前主要是通过ERK1/2相关信号通路活性调节肠上皮损伤愈合能力以及通过钙离子相关信号通路招募巨噬细胞等单核细胞迁移到炎症部位[16,27]。而与之相对应的GPR35下游包括Gi/o,Gq在内的小G蛋白激活以及β-arrestin招募相关[20,28,29]。本研究通过体外构建GPR35、GPR35T108M、GPR35S294R表达载体,利用钙流检测,β-arrestin招募实验以及CRE荧光素酶报告系统对其进行受体活性分析,最终证明rs3749171,rs3749172位点突变会导致GPR35受体活性增强,下游相关信号通路激活。最新的关于在结肠癌的研究中也证明了GPR35-T108M突变增强了GPR35调控下游Na/K-ATP酶的活性的能力[30]。结合前面的实验结果,本研究能够得到最终结论:肠炎相关SNP位点rs3749171和rs3749172能够增强GPR35受体活性及下游相关信号通路进而促进肠炎的发生发展。而这个结果也与前文敲除动物模型结果一致,最终从动物模型及病人SNP相关易感群体分析两个方向共同验证了在肠炎发生发展中的作用。
ERK1/2信号通路是GPCR下游重要的信号通路组成部分,能够调控细胞增殖,凋亡及迁移等细胞进程[20]。而对敲除小鼠肠炎模型肠上皮WB分析发现:相较于野生型小鼠,敲除后,小鼠结肠上皮细胞中ERK1/2磷酸化水平在肠炎发生过程中显著上调。这说明能够通过抑制ERK1/2信号通路的方式促进肠炎的发生发展。在肠炎病理进程中,肠上皮ERK1/2相关信号通路能够通过调控肠上皮粘膜伤口愈合的方式抑制肠炎的发生发展[16,31]。这些研究与DSS肠炎模型中敲除小鼠肠上皮ERK1/2磷酸化水平上调的结果保护肠炎相关论证匹配。实验结果与前人研究基础相结合证明了能够通过抑制下游ERK1/2相关信号通路活性的方式促进肠炎的发生发展。
在肠炎发生发展进程中,肠道菌群在其中起到了重要的作用。肠道菌群的多种代谢产物通过肠上皮细胞GPR43,TLR4等受体正向或负向调节肠上皮通透性及免疫反应强度,而肠道免疫系统则通过分泌抗菌肽,调节自身免疫活性的方式维持肠道内菌群稳定及肠道上皮通透性等生理状态[32~34]。肠道菌群失衡及机体应对肠道菌群免疫反应失衡均会导致肠炎的发生发展[35]。在肠上皮细胞中高表达且能够识别犬尿酸在内的多种细菌代谢产物,调控下游相关信号通路活性变化[36,37]。这说明在肠道系统中能够通过监测各类细菌代谢产物浓度变化的方式调控下游信号通路激活/抑制程度,进而调节肠上皮通透性及下游免疫系统活性。在发生SNP相关突变后,GPR35识别犬尿酸等代谢物配体能力及激活下游信号通路的能力增强,并持续促进/抑制下游肠上皮细胞及免疫细胞抵抗细菌入侵的进程。在发生肠上皮损伤或菌群失调等特殊情况下,GPR35相关信号通路异常活化会导致免疫系统与菌群的平衡更容易被破坏,大概率提升肠炎发病几率及肠炎严重程度。
综上所述,本研究初步验证了肠炎相关SNP位点rs3749171和rs3749172能够上调GPR35受体活性,且GPR35激活能够促进DSS诱导的肠炎的发生发展。上述结果将为后续肠炎发生病机制及后续肠炎治疗药物的开发提供新的靶点。
[1] Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease, 2007, 448(7152): 427–34.
[2] Lewis JD, Deren JJ, Lichtenstein GR. Cancer risk in patients with inflammatory bowel disease1999, 28(2): 459–477.
[3] Itzkowitz SH, Yio XY. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation, 2004, 287(1): G7–17.
[4] Konda A, Duffy MC. Surveillance of patients at increased risk of colon cancer: inflammatory bowel disease and other conditions, 2008, 37(1): 191–213.
[5] Ananthakrishnan AN, CN Bernstein, D Iliopoulos, A Macpherson, MF Neurath, RAR Ali, SR Vavricka, C Fiocchi, Environmental triggers in IBD: a review of progress and evidence., 2018. 15(1): 39–49.
[6] Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, Vavricka SR, Fiocchi C. Environmental triggers in IBD: a review of progress and evidence, 2018, 15(1): 39–49.
[7] Liu TC, Stappenbeck TS. Genetics and pathogenesis of inflammatory bowel disease, 2016, 11: 127–148.
[8] Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, Latiano A, Goyette P, Green T, Halfvarson J, Haritunians T, Korn JM, Kuruvilla F, LagacéC, Neale B, Lo KS, Schumm P, Törkvist L, Dubinsky MC, Brant SR, Silverberg MS, Duerr RH, Altshuler D, Gabriel S, Lettre G, Franke A, D'Amato M, McGovern DP, Cho JH, Rioux JD, Xavier RJ, Daly MJ. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease, 2011, 43(11): 1066–1073.
[9] Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, Kugathasan S, Bradfield JP, Walters TD, Sleiman P, Kim CE, Muise A, Wang K, Glessner JT, Saeed S, Zhang H, Frackelton EC, Hou C, Flory JH, Otieno G, Chiavacci RM, Grundmeier R, Castro M, Latiano A, Dallapiccola B, Stempak J, Abrams DJ, Taylor K, McGovern D, Silber G, Wrobel I, Quiros A, Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmuda MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwillam R, Tremelling M, Delukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ, Heyman MB, Ferry GD, Kirschner B, Lee J, Essers J, Grand R, Stephens M, Levine A, Piccoli D, Van Limbergen J, Cucchiara S, Monos DS, Guthery SL, Denson L, Wilson DC, Grant SF, Daly M, Silverberg MS, Satsangi J, Hakonarson H. Common variants at five new loci associated with early-onset inflammatory bowel disease, 2009, 41(12): 1335–1340.
[10] Ellinghaus D, Folseraas T, Holm K, Ellinghaus E, Melum E, Balschun T, Laerdahl JK, Shiryaev A, Gotthardt DN, Weismüller TJ, Schramm C, Wittig M, Bergquist A, Björnsson E, Marschall HU, Vatn M, Teufel A, Rust C, Gieger C, Wichmann HE, Runz H, Sterneck M, Rupp C, Braun F, Weersma RK, Wijmenga C, Ponsioen CY, Mathew CG, Rutgeerts P, Vermeire S, Schrumpf E, Hov JR, Manns MP, Boberg KM, Schreiber S, Franke A, Karlsen TH. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4, 2013, 58(3): 1074–1083.
[11] Yang SK, Hong M, Oh H, Low HQ, Jung S, Ahn S, Kim Y, Baek J, Lee CH, Kim E, Kim KM, Ye BD, Kim KJ, Park SH, Lee HS, Lee I, Shin HD, Han B, McGovern DP, Liu J, Song K. Identification of loci at 1q21 and 16q23 that affect susceptibility to inflammatory bowel disease in koreans, 2016, 151(6): 1096–1099.e4.
[12] Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation, 2001, 29(1): 308–311.
[13] King N, Hittinger CT, Carroll SB. Evolution of key cell signaling and adhesion protein families predates animal origins, 2003, 301(5631): 361–363.
[14] Mackenzie AE, Lappin JE, Taylor DL, Nicklin SA, Milligan G. GPR35 as a novel therapeutic target, 2011, 2: 68.
[15] Zheng X, Hu MM, Zang XJ, Fan QL, Liu YL, Che Y, Guan XJ, Hou YL, Wang GJ, Hao HP. Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress, 2019, 79: 244–255.
[16] Tsukahara T, Hamouda N, Utsumi D, Matsumoto K, Amagase K, Kato S. G protein-coupled receptor 35 contributes to mucosal repair in mice via migration of colonic epithelial cells, 2017, 123: 27–39.
[17] Zeng Z, Mukherjee A, Varghese AP, Yang XL, Chen S, Zhang H. Roles of G protein-coupled receptors in inflammatory bowel disease, 2020, 26(12): 1242–1261.
[18] Horii Y, Uchiyama K, Toyokawa Y, Hotta Y, Tanaka M, Yasukawa Z, Tokunaga M, Okubo T, Mizushima K, Higashimura Y, Dohi O, Okayama T, Yoshida N, Katada K, Kamada K, Handa O, Ishikawa T, Takagi T, Konishi H, Naito Y, Itoh Y. Partially hydrolyzed guar gum enhances colonic epithelial wound healingactivation of RhoA and ERK1/2, 2016, 7(7): 3176–3183.
[19] Hattori M, Tanaka M, Takakura H, Aoki K, Miura K, Anzai T, Ozawa T. Analysis of temporal patterns of GPCR-β-arrestin interactions using split luciferase-fragment complementation, 2013, 9(5): 957–964.
[20] Carmona-Rosas G, Alcántara-Hernández R, Hernández- Espinosa DA. Dissecting the signaling features of the multi-protein complex GPCR/β-arrestin/ERK1/2, 2018, 97(5): 349–358.
[21] Bennet JD, Brinkman M. Treatment of ulcerative colitis by implantation of normal colonic flora, 1989, 1(8630): 164.
[22] Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, Talley NJ, Moayyedi P. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis, 2011, 106(4): 661–673.
[23] Feagan BG, Rochon J, Fedorak RN, Irvine EJ, Wild G, Sutherland L, Steinhart AH, Greenberg GR, Gillies R, Hopkins M. Methotrexate for the treatment of Crohn's disease. The north american crohn's study group investigators, 1995, 332(5): 292–297.
[24] Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there?, 2006, 5(12): 993–996.
[25] Milligan G. Orthologue selectivity and ligand bias: translating the pharmacology of GPR35, 2011, 32(5): 317–325.
[26] Quon T, Lin LC, Ganguly A, Tobin AB, Milligan G. Therapeutic opportunities and challenges in targeting the orphan G protein-coupled receptor GPR35, 2020, 3(5): 801–812.
[27] Maravillas-Montero JL, Burkhardt AM, Hevezi PA, Carnevale CD, Smit MJ, Zlotnik A. Cutting edge: GPR35/CXCR8 is the receptor of the mucosal chemokine CXCL17, 2015, 194(1): 29–33.
[28] Chen M, Cecon E, Karamitri A, Gao W, Gerbier R, Ahmad R, Jockers R. Melatonin MT1and MT2receptor ERK signaling is differentially dependent on Gi/oand Gq/11proteins, 2020, 68(4): e12641.
[29] Bonder DE, McCarthy KD. Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex, 2014, 34(39): 13139–13150.
[30] Schneditz G, Elias JE, Pagano E, Zaeem Cader M, Saveljeva S, Long K, Mukhopadhyay S, Arasteh M, Lawley TD, Dougan G, Bassett A, Karlsen TH, Kaser A, Kaneider NC. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump, 2019, 12(562): eaau9048.
[31] Zhao PW, Sharir H, Kapur A, Cowan A, Geller EB, Adler MW, Seltzman HH, Reggio PH, Heynen-Genel S, Sauer M, Chung TD, Bai Y, Chen W, Caron MG, Barak LS, Abood ME. Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity, 2010, 78(4): 560–568.
[32] Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases, 2019, 10: 277.
[33] Melhem H, Kaya B, Ayata CK, Hruz P, Niess JH. Metabolite-Sensing G protein-coupled receptors connect the diet-microbiota-metabolites axis to inflammatory bowel disease, 2019, 8(5): 450.
[34] Dejban P, Nikravangolsefid N, Chamanara M, Dehpour A, Rashidian A. The role of medicinal products in the treatment of inflammatory bowel diseases (IBD) through inhibition of TLR4/NF-kappaB pathway, 2020.
[35] Alam MT, Amos GCA, Murphy ARJ, Murch S, Wellington EMH, Arasaradnam RP.S Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels, 2020, 12: 1.
[36] Deng HY, Hu HB, Fang Y. Multiple tyrosine metabolites are GPR35 agonists, 2012, 2: 373.
[37] Wang JH, Simonavicius N, Wu XS, Swaminath G, Reagan J, Tian H, Ling L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35, 2006, 281(31): 22021–22028.
Inflammatory bowel disease susceptible genepromotes bowel inflammation in mice
Yansen Zheng, Lingang Zhuo, Dali Li, Mingyao Liu
Inflammatory bowel disease (IBD) has emerged as a public health challenge with high incidence, recurrence rates and low cure rate. Moreover, sustained inflammation increases the risk of colorectal cancer. The occurrence and progression of IBD are closely related to the genetic mutation. Previous genome-wide association studies (GWAS) analysis demonstrated that the susceptibility loci rs4676410, rs3749171, and rs3749172 in thegene locus increase the risk of IBD, but no direct evidence on the function ofin IBD progression has been shown. To investigate the role ofin IBD, CRISPR/Cas9 technology was employed to construct aknockout mouse strain. TheGpr35mice exhibited lower susceptibility to dextran sodium sulfate-induced IBD model than the wildtype group with a significant reduction in body weight loss, DAI score, intestinal epithelial injury, and macrophage cell infiltration. To explore how the IBD susceptibility loci rs3749171 and rs3749172 regulate GPR35 activity, two mutant forms of GPR35 (T108M and S294R) were constructed. By analyzing the activity of GPR35 downstream signaling pathway, the two mutation forms of GPR35 exhibited higher receptor activity to Zaprinast than the wildtype GPR35. Finally, the Western blotting analysis found an elevated phosphorylation level of ERK1/2 inGpr35colon epithelial after DSS treatment, demonstrating that the loss function ofalleviates the IBD syndrome by activating the ERK1/2 signaling pathway. In summary, the IBD susceptibility loci rs3749171 and rs3749172 may promote the disease progression by activating GPR35 activity, providing a potential drug target for the treatment of inflammatory bowel disease.
rs3749171; rs3749172;; IBD; animal model
2020-11-18;
2020-12-31
国家重点研发计划基金项目(编号:2019YFA0110802)和国家自然科学基金面上项目(编号:81670470, 81873685)资助[Supported by grants from National Key R&D Program of China (No. 2019YFA0110802), and the National Natural Science Foundation of China (Nos. 81670470, 81873685)]
郑燕森,在读博士研究生,专业方向:肠炎分子机制研究。E-mail: zhengysen@126.com
李大力,博士,教授,研究方向:动物模型构建新技术开发及基因编辑技术在基因治疗中的应用。E-mail: dlli@bio.ecnu.edu.cn刘明耀,博士,教授,研究方向:G-蛋白偶联受体(GPCRs)及其信号转导途径在疾病发生过程中的作用。E-mail: myliu@bio.ecnu.edu.cn
10.16288/j.yczz.20-392
2021/1/28 8:34:48
URI: https://kns.cnki.net/kcms/detail/11.1913.r.20210127.1133.001.html
(责任编委: 卢大儒)
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13
天津医科大学学报(2021年4期)2021-08-21
中国民间疗法(2020年22期)2021-01-07
中国民族医药杂志(2016年2期)2016-05-14
中国民族医药杂志(2016年5期)2016-05-09
中国癌症杂志(2015年4期)2015-12-09
实用手外科杂志(2015年3期)2015-08-27
中国卫生标准管理(2015年3期)2015-01-27
西南军医(2015年2期)2015-01-22
当代畜禽养殖业(2014年5期)2014-08-31
