
基因解读系统中遗传检测报告自动生成技术
2021-02-25 07:48张少伟蒋艳凰
计算机应用与软件 2021年2期
张少伟 蒋艳凰
1(中国科学技术大学软件工程学院 安徽 合肥 230026)2(人和未来生物科技(长沙)有限公司 湖南 长沙 410000)
0 引 言
遗传病是指染色体畸变和基因突变引起的一大类疾病。截至2010年8月10日,已登记的孟德尔遗传性状或者疾病达20 135种,其中已知的单基因遗传病及线粒体基因病为6 500余种[1]。由于我国人口基数庞大,遗传病对我国的影响尤为明显,我国有1 000余万单基因遗传病患者,新生儿中有超过800万染色体遗传病患者,多基因遗传病和体细胞遗传病的发病人数更是难以估量[2]。对基因进行精准检测与解读,准确定位致病原因,预防遗传疾病,成为一项关系到全人类的关键技术,也是近年来临床医学研究的热点。
外显子测序是指用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法[3]。在人类基因中大约有180 000外显子,一个正常样本的基因检测结果可能包含了数万条突变基因,由人工进行逐条解读是不实际也是不合理的。在这一背景下,国内外推出了众多优秀的基因解读系统,如“人和未来”的GTX.Digest系统、以色列的Congenica等。这些系统能够对基因进行注释并按致病性排序,医疗人员仅需要关注排名前几十个基因,极大地减少了医疗工作者的工作量。医疗工作者依据解读系统的分析结果,查找一定量的文献来佐证基因与疾病之间的影响,并最终生成一份解读报告。
中国遗传学会遗传咨询分会组织发布的《高通量全外显子组测序检测报告示例》中显示,检测报告应包含样本信息、临床症状、检测项目、检测结论、基因变异信息、疾病名称、结果解释、建议,以及参考文献[4]。这样一份报告往往需要花费医疗工作者数小时的工作时间,一些复杂的报告甚至需要花费数十小时。因此,自动出具报告并将已有信息自动填入报告能够极大减轻医疗工作者的负担。
GTX.Digest是“人和未来”推出的一款云端基因解读系统。GTX.Digest以基因突变为单位,以探究突变的致病性为目标,对用户的VCF数据文件进行注释解读。解读结果不仅包含基因数据的注释结果,还包含ClinVar、OMIM、Orphanet等权威数据库的致病性建议。同时,GTX.Digest开发组还对PubMed所有文献进行了文本挖掘,探究文献描述的基因突变与疾病之间的关系,并开创性地将文本挖掘结果应用在了基因解读中,用户可直接看到文献中描述基因突变与疾病关系的句子,极大地提高了医生确定致病基因的效率和准确性。GTX.Digest能够很好地辅助医生进行遗传病分析,但无法自动生成遗传检测报告,而遗传检测报告作为基因解读的最终输出,是一个成熟的基因解读系统中不可或缺的一环[5]。
针对自动出具报告的现实需求,基于GTX.Digest基因解读系统的分析结果、医生对致病性的判定结果、生物医学领域的权威数据库等,在Linux操作系统上,实现了全外显子检测报告的自动生成。用户在解读系统中确定了致病基因后,填写患者基本信息,选择与治病基因相关联的疾病、转录本等信息,系统将自动分析基因变异产生的氨基酸变化,自动生成从基因便宜角度对疾病解释,以及与之相关的参考文献。系统提供报告预览和自动下载功能,下载的报告为Word格式,用户可对其进行进一步编辑。
1 报告自动生成系统架构
图1给出了报告自动生成系统结构,主要包括三个部分:基因数据库、报告内容获取、报告生成与应用。基础数据库包括生成报告所需的基础数据来源,这些数据包括:GTX.Digest系统解读结果、氨基酸描述数据、表型-疾病关系数据、文献数据等。数据的真实性与权威性决定了报告的质量,因此必须以准确、权威的数据库为基础,构建报告生成系统。报告内容的获取包括两部分:一是需要医护人员参与的内容获取,包括填写用户的基因信息、从解读结果中选择致病基因相关的疾病和转录本等;二是系统根据基础数据库自动生成相关报告内容。报告生成和应用则根据获取的情报内容,自动生成Word报告。

图1 报告自动生成系统结构
1.1 基础数据库
基础数据库是报告自动生成的关键,决定了报告的整体质量,因此数据来源必须与行业规范相符合,数据的权威性也需得到行业的认可。
1.1.1解读结果数据
解读结果是指对原始的基因数据的解读结果,将生涩难懂的基因编码转换成普适的学科术语,并以基因突变为单位,依据权威数据库,对其致病性进行标注[6]。
该系统的解读结果数据来源于基因解读平台GTX.Digest。GTX.Digest不仅对基因和变异进行了致病性排序,而且展示了对PubMed文献的挖掘结果,使得解读结果更科学、精准。
解读结果数据包括:基因名、转录本编号、突变信息、遗传模式、致病性、OMIM疾病等。
1.1.2氨基酸描述数据
解读结果数据中关于氨基酸的变化使用的是国际通用的氨基酸简写形式,而在报告中是以中文形式呈现。氨基酸描述数据记录了氨基酸的英文简称与中文全称,用来实现英文简称与中文全称的转换,如表1所示。

表1 氨基酸描述数据
1.1.3表型-疾病对应数据
表型指一定基因型的个体,在特定环境中所呈现出来的性状,例如:感冒、发烧、红发[6]。所有的遗传病都有其特定的表型特征,正是因为表型与疾病的这种相关性,解读软件要求输入表型数据来作为排序依据。
通常来说,基因突变可能导致的疾病不唯一,每种疾病的表型特征也不尽相同[7]。要精确判定样本患有何种疾病,必须计算样本表型与疾病的相关性,这就需要表型-疾病关系数据,即某一种疾病会有哪些表型。
人类孟德尔遗传线上库(Online Mendelian Inheritance in Man,OMIM)[8]提供了疾病与人类表型术语集(Human Phenotype Ontology,HPO)的对应表,如表2所示。

表2 OMIM疾病-HPO对应关系
OMIM提供的表格以OMIM疾病为基本单位,每一个疾病-表型关系为一条,共96 919条数据。在本文系统中,疾病-表型关系用于判定用户输入的表型与何种疾病相关性更大,这种相关性用“用户输入表型”与“疾病对应表型”相同的数量来衡量。依据这种应用场景,对表格进行了聚合,得到7 015条MySQL记录,结构如表3所示。

表3 聚合后的OMIM疾病-表型对应
为方便对HPO进行比对,以列表风格对HPO进行存储,同时为了加快查询速度,本文还对OMIM编号添加了索引。
1.1.4文献数据
遗传检测报告用于指导医生进行临床诊断,因此报告的内容,特别是致病性判定一定要有所依据,需要一定量的参考文献支持。
本系统中的参考文献来源于权威数据库OMIM、ClinVar、PubMed文献挖掘结果库dmVar,并在其基础上进行了优化排序。
1.2 报告内容获取
报告内容获取即生成报告内容,主要分为用户基本信息填写、致病基因分析结果获取、致病性描述、文献获取四个部分。
(1) 用户基本信息填写依据《高通量全外显子组测序检测报告示例》。报告中的基本信息应包括受检者信息、样本信息、送检者信息,由用户填写。此外,用户还可对系统推荐的转录本和疾病进行更改。
(2) 致病基因分析结果包括基因名、转录本、遗传方式等信息。从GTX.Digest解读结果中获取数据,并按照相应的表格形式进行组织。
(3) 致病性描述中除了解读结果外,还包括基因所导致的疾病、疾病遗传方式、父母的患病分析。
(4) 文献获取指从文献数据库中获取疾病所对应的参考文献,并按照参考文献格式进行组织。
1.3 报告生成与应用
全外显子检测报告包含表格类复杂结构,目前没有很好的Linux库可实现对Word的直接编辑。为了兼顾Web页面预览的需求,本文决定使用HTML作为直接生成格式,而后再对其进行格式转换。
在报告生成过程中存在多次页面交互:基本信息填写与疾病转录本选择、疾病-基因相关性检查、报告预览。
在基本信息填写与疾病转录本选择交互界面中,用户可填写基本信息,还可以更改系统推荐的疾病与转录本。用户填写的基本信息将进行存储,用于自动填充信息表格。
当用户选择的疾病与基因的遗传方式不同时,相关数据将呈现在疾病-基因相关性检查界面上,供用户查看和确认。
HTML报告生成后,可进行报告预览。
2 HTML模板生成与文献排序
遗传报告自动生成的难点主要有两点:Linux系统下Word文件的生成和文献数据排序。在本技术中,Word报告的生成路线为:HTML模板—数据填充—格式转换,HTML模板定义了报告的内容和风格,是报告生成的关键。文献数据是报告结论可信度的重要依据,由于报告的篇幅有限,如何对文献进行排序,并从中选出可信度大的文献,是提高报告质量的关键。
2.1 HTML模板的生成
模板指的是报告的初始HTML模板,其定义了报告的基本结构。模板由Word类软件编辑并导出,而后根据所填写的内容,对模板内容进行调整。模板内容主要分为4类:固定表格、自由表格、固定段落、自由段落,每一类都有各自的处理方法。
(1) 固定表格。固定表格的特征是表格结构不会随报告内容改变而改变,如表4所示。因此,表格的格式可以固定在模板中,只需要将填入的信息进行替换即可。

表4 固定表格
(2) 自由表格。自由表格指的是表格的结构会随报告的内容而改变。例如表5所示的临床表型相关变异表格。

表5 自由表格-临床表型相关变异
表格的大小会随着用户标记的基因数量而增加,因此表格的结构不能够固定在模板中,而是应该随着数据的增加,将结构连同数据一同写入。因此,在HTML模板中,临床表型相关变异表格仅写入表头格式。基因数据按照表头格式,逐行进行插入。
(3) 固定段落。固定段落的内容一般是通用的解释性语句,语句内容固定,格式固定,可直接定义在模板中。
(4) 自由段落。自由段落指段落的内容不固定,其中又分为字符不固定段落与格式不固定段落。字符不固定段落指的是段落的基本格式已经确定,仅有段落中的某些字符串需要随着用户的输入而进行更改,这类段落可以直接编写在HTML模板中,将需要替换的关键字进行标识,使用时进行字符替换。
格式不固定段落指的是段落格式或数量不固定,这类段落无法将格式固定在HTML模板中,故HTML模板仅写入需要替换的字符串,段落格式由内容生成模块定义。
2.2 文献排序
基因检测报告需要提供PubMed文献的引用作为文献,如何获取与基因检测报告内容相关的文献成为关键。本文利用了三个数据来源:OMIM数据库、ClinVar数据库和文本挖掘数据库dmVar。
OMIM数据库是人类孟德尔遗传的网络版,主要着眼于可遗传的或遗传性的基因疾病,其中包括文本信息和相关参考信息、序列记录、图谱和相关其他数据库[8]。
ClinVar是一个公开的数据库,收集了与疾病相关的数据库。由美国国立卫生研究院于2013年为了生物技术信息开发而构建。ClinVar拥有来自1 000个提交者的600 000条提交记录,代表430 000条变异数据[9]。
PubMed是由美国国家生物技术中心(NCBI)主导的,美国国立卫生研究院(NIH)与美国国家医学图书馆(NLM)共同开发维护的免费数据库,其收录了3 000万篇生物医学文献[10]。PubMed文献的挖掘工作一直是医学工作者的研究热点,在进行挖掘时,研究者主要采用3种方法将描述表型的词汇映射为标准词表[11],包括医学主题词(Medical Subject Headings, MeSH)[12]、统一医学语言系统(Unified Medical Language System, UMLS)[13]、人类表型本体论(Human Phenotype Ontology, HPO)[14]。GTX.Digest系统中的文本挖掘数据库dmVar以MeSH号作为表型标准词表,提取了PubMed文献中基因、突变、疾病等命名实体,以及期刊编号、期刊影响因子等信息。
基于上述三个数据库,本文选择文献的优先级判断主要有相关性和可信度两个维度。相关性指文献是否在描述所选基因和疾病,是一个强条件;可信度是一个较为综合的指标,以期刊的影响因子和发表时间进行综合评估。
2.2.1文献预处理
ClinVar和OMIM虽然都是遗传疾病的数据库,但两者的关注点不一样。ClinVar以基因突变为基本单位,探讨突变是否致病,寻找致病性证据。OMIM则是以疾病为基本单位,讨论疾病由哪些基因突变引起,给出疾病的基本特征。在这一差异下,两者文献数据的组织方法有很大差别。OMIM提供的文献列表明确了文献讨论的基因与疾病;ClinVar给出的文献列表则关注突变及其是否会致病,没有对疾病进行分类。相关性(文献是否描述同一个疾病)是进行文献排序的前提,因此需要对ClinVar文献进行分类。此外,两者都没有给出文献所发期刊的影响因子。
文献预处理的主要工作有:(1) 实现ClinVar文献按疾病分类;(2) 获取文献影响因子标记。
2.2.2ClinVar文献分类
dmVar文本挖掘数据库中标注了每一篇文献所讨论的基因、突变、疾病、期刊影响因子,其中疾病以PubMed疾病分类标准MeSH进行标注。
基于文本挖掘结果,ClinVar文献预处理算法流程如图2所示。使用ClinVar文献的PubMed号在文本挖掘结果中查找该文献所讨论疾病的MeSH号,根据查询到的MeSH号,于MeSH-OMIM号对应表中查找MeSH号所对应的OMIM号,最后以此OMIM号来标记ClinVar文献,实现ClinVar文献的疾病分类。
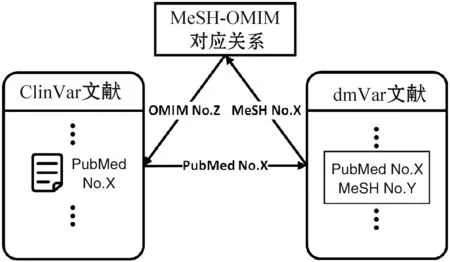
图2 ClinVar文献分类流程
2.2.3期刊影响因子的获取
PubMed文本挖掘结果中包含了文献所发期刊的影响因子,由于PubMed收录文章的跨度大(1781年至今),一些期刊已经停刊或更名,对于这类期刊,本文默认其影响因子为0。
期刊影响因子的获取如图3所示。使用文献的PubMed号在文本挖掘结果中查询对应期刊的影响因子,并以影响因子标记文献。

图3 期刊影响因子获取流程
2.2.4排序算法
文献排序的评定标准主要有:相关性、影响因子、发表时间。
1) 相关性判断。实现ClinVar文献的疾病分类后,依据疾病可获得OMIM文献列表和ClinVar文献列表集合。集合内的文献与所述疾病绝对相关,应当拥有更高的权重,否则其权重应当较小。
2) 影响因子的权重设计。影响因子IF是以年为单位进行计算的。以1992年的某一期刊影响因子为例:
IF1992=A/B
式中:A为该期刊1990年至1991年所有文献在1992年中被引用的次数;B为该期刊1990年至1991年所有文献数。文献刊物的影响因子来源于汤森路透发布的期刊引证报告(Journal Citation Reports,JCR)[15],其代表最近一年内刊物的论文质量。据此,文献发表时间越早,当前影响因子的有效性应当越低;另一方面,随着检测设备和现代医学的发展,发表时间晚的文献应当具有更高的可信度,其影响因子也应该有更高的权重。
本文统计了2002年至2012年遗传学排名前9的期刊的影响因子变化率,如表6所示。可以看出,排名前9的期刊影响因子的10年平均变化率高达35%,证明了影响因子权重设计的正确性。文献发表时间越早,当前影响因子的有效性就越低。

表6 遗传学期刊影响因子变化率
基于上述思想,设计排序算法如下:
pscore=α·r·EIF(IF+0.01)+(1-α)·(1-r)·EIF(IF+0.01)
式中:r为相关性,表示文献是否与疾病主题相关,相关时r=1,否则r=0;α为相关性因子,取α=0.99;IF为当前年份期刊的影响因子,(IF+0.01)是为了避免未找到影响因子的文献被直接排除;EIF为影响因子的时效性量化。
式中:Yc为所有文献发表时间的中位数,即对发表时间越早的文献,当前年份影响因子的影响力越低。
3 实 验
文献排序算法的目标是找到符合普遍医学研究者文献选取倾向的文献序列,通常来说依据有:(1) 主题一致;(2) 影响因子大;(3) 发表时间近。
3.1 单维算法结果分析
基于上述的基本依据,考察相关性、影响因子、发表时间各自对算法结果的影响。本文假设文献列表中文献发布时间中位数为1970年,即:
分别考察(r=0,IF=10)、(r=1,IF=10)、(r=1,IF=15)条件下,Pscore随发表时间的变化规律,结果如图4所示。

图4 不同条件下Pscore随发表时间的变化曲线
依据变化曲线结果,从相关性、影响因子、发表时间三个维度进行分析,可得:
1) 由P1、P2曲线对比可知,在本文算法的排序结果中,相同发表时间、相同影响因子、主题相关的文献比不相关的文献的排序结果要靠前。
2) 由P2、P3曲线对比可知,在本文算法的排序结果中,主题相关的、发表时间相同的、较高影响因子的文献的排序要优于较低影响因子的文献。
3) 由P3曲线可知,在本文算法的排序结果中,主题相关的、影响因子相同的、发表时间较晚(较新)的文献的排序要优于较早发表的文献。
由上述的分析结果可知,本文排序算法符合医学研究者普遍的文献排序规则,排序结果符合用户的预期。
3.2 多维算法结果分析
在真实的使用环境中,单维度影响是容易决断的,难的是多维度分析。例如此时有两篇文献A、B,其中A发表于2002年,当前影响因子为4,B发表于2007年,当前影响因子为3.5,此时A、B的排序是难以确定的,抉择时应该考虑使用场景。此时,存在两种不同的场景:候选文献的发表时间普遍较早(场景一)和候选文献的发表时间普遍较晚(场景二)。
在场景一中,文献的发表时间都较早,发表时间影响力降低,影响因子为主要判断因素(没有充分的可供选择“新”文献),此时A文献的排序应该高于B。在场景二中,文献的发表时间都较晚,早发表的文献的优先级降低(有充分的可供选择的“新”文献),此时B文献的排序应该高于A。
依据上述假设,引入文献列表发表时间中位数Yc,假定两篇文献分别为T1(Yc=2010,IF=10)、T2(Yc=1990,IF=15) ,其中Yc为发表时间、IF为影响因子。考察中位数对算法结果的影响,如图5所示。

图5 中位数Yc-Pscore曲线
可以看出,当中位数较低时,T2排序结果优于T1,即影响因子为主要影响因素;当中位数较高时,T1的排序结果优于T2,此时影响因子的影响力降低,发表时间影响力增加。该结果符合引入中位数的初衷。
3.3 排序实验
本实验选取了OMIM编号为216900疾病对应的17篇文献,并使用本文算法对其进行排序,结果如表7所示。

表7 遗传学期刊影响因子变化率

续表7
可以看出,本文算法并不是单一从影响因子或者发表时间进行排序,而是对二者进行了复合考虑,排序结果符合我们的预期。
3.4 算法评价
文献排序算法的最终目标是从特定的文献列表中,选出可信度高的文献,这一过程应该考虑文献列表的特点,而不是单一地使用某一项指标衡量。本文算法不仅综合了相关性、影响因子和发表时间的影响,还考虑了文献列表整体发表时间的影响,能够满足实际应用的需求。
4 结 语
本文从医生出具遗传检测报告过于繁琐的现实需求出发,实现了遗传检测报告自动生成技术。该技术整合了OMIM、ClinVar、PubMed文献挖掘数据dmVar,并在其基础上设计了文献排序算法。系统可针对用户输入的表型,推荐匹配度更高的遗传疾病,提高了检测报告的准确性,减轻了医生的工作量。遗传检测报告自动生成技术已经应用在GTX.Digest中。下一步工作为:(1) 拓展数据库,加入其他权威数据库如Orphanet等。(2) 拓展系统可出具报告的类型,如肿瘤检测报告等。(3) 在报告中加入疾病描述的内容。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
农村科学实验(2022年2期)2022-03-12
名家名作(2021年4期)2021-05-12
科普童话·学霸日记(2020年1期)2020-05-08
三农资讯半月报(2020年2期)2020-03-09
小天使·一年级语数英综合(2019年2期)2019-01-10
华人时刊(2018年17期)2018-11-19
数学学习与研究(2018年7期)2018-05-16
山东青年(2017年11期)2018-03-29
都市丽人(2015年4期)2015-03-20
