
三元混配铜(Ⅱ)配合物的晶体结构、DNA作用及其生物活性
2021-02-24 00:48:52蔡戴宏莫慧雯乐学义
无机化学学报 2021年1期
蔡戴宏 莫慧雯 何 良 乐学义
(华南农业大学材料与能源学院应用化学系,广州 510642)
肿瘤细胞具有无限增殖、分化以及易扩散等共同特征,目前临床上广泛应用顺铂、卡铂等铂类配合物进行治疗。然而,这类药物存在交叉耐药性、对某些肿瘤细胞活性较低、体内不易代谢和严重毒副作用等缺点,因此新型、高效的金属配合物药物成为热门研究领域,尤其是低毒的内源性金属配合物吸引了人们的关注[1-4]。铜(Ⅱ)离子是重要的内源性金属,具有很强的配位能力,能和大多数芳杂环(含N、S、O、P)有机化合物配位,形成结构复杂、配位模式多变的金属铜(Ⅱ)配合物。同时,研究发现铜(Ⅱ)配合物与铂类配合物不同,主要以非共价结合的方式作用于DNA,这种结合方式可以提供更广泛的抗肿瘤活性[5-7]。2-取代苯并咪唑衍生物具有一定生物活性,可作为配体形成铜(Ⅱ)配合物,并且可通过芳香环上引入不同官能团进一步提高和调节金属配合物的生物活性。氨基酸为重要的生命内源物质,作为辅助配体与金属元素形成配合物时,不仅能够提高配合物的生物活性,而且能够降低配合物的毒副作用[8-11]。
基于以上原因,我们以5-氯-2-(2'-吡啶基)苯并咪唑(5-chloro-2-(2'-pyridyl)benzimidazole,HPBC)为主配体、L-苯丙氨酸根(L-phenylalaninate,L-Phe)为辅助配体,合成以铜(Ⅱ)为中心离子的配合物:[Cu(HPBC)(L-Phe)(H2O)]ClO4(1),并采用元素分析、摩尔电导率和X射线单晶衍射等方法确定了配合物的组成和结构。重要的是,研究了配合物的DNA结合性质与生物性质:如通过电子吸收光谱、荧光光谱、粘度实验以及分子对接技术研究了配合物与DNA的相互作用方式;分别采用牛津杯法和噻唑蓝(3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium,MTT)比色法测定配合物对细菌及肿瘤细胞生长的抑制作用,并通过吖啶橙(acridine orange,AO)/溴化乙锭(ethidium bromide,EB)染色法、流式细胞仪测定细胞周期等方法初步揭示了其抗肿瘤作用机制。
1 实验部分
1.1 材料与仪器
L-苯丙氨酸、小牛胸腺DNA(calf thymus DNA,CT-DNA)、三羟基氨基甲烷(tris(hydroxymethyl)aminomethane,Tris)、溴化乙锭(ethidium bromide,EB)、牛肉浸膏、细菌学蛋白胨、琼脂粉、DMEM培养基(dulbecco's modified eagle medium,DMEM)、RPMI-1640培养基(RPMI 1640 medium,RPMI-1640)、胎牛血清、新生牛血清、胰蛋白酶、核糖核酸酶A(Ribonuclease A,RNAse)、碘化丙啶(propidium iodide,PI)均为生化试剂;其他试剂均为市售,纯度为分析纯。
所用仪器有:DDS-11A数显电导率仪,上海雷磁新泾仪器有限公司;ACATAR 360 FT-IR型红外光谱仪,美国Nnicolet公司;Vario EL Cube元素分析仪,德国Elementar公司;Pharmacia UV-2550紫外-可见分光光度计,日本Shimadzu公司;F-7000荧光光谱仪,日本Hitachi公司;API 3200液质联用仪,AB Sciex公司;MCO-15AC型二氧化碳培养箱,日本松下电器公司;CKX31-A11RC型倒置显微镜,Olympus公司;Varioskan Flash型多功能酶标仪,美国Thermo Scientific公司;ImageXpress Micro XLS高内涵成像分析系统,Molecular Devices公司;FACS Calibur流式细胞仪,美国BD Bioscience。
1.2 配合物1的合成
根据文献[12]中的方法制备配体HPBC。
配合物1的合成:将Cu(ClO4)2水溶液(0.5 mmol,0.504 0 mol·L-1)滴加到含有 NaOH(0.5 mmol,0.020 0 g)的L-苯丙氨酸(0.5 mmol,0.082 6 g)水溶液(5 mL)中,在50℃下搅拌反应20 min。随后,滴加含有HPBC(0.5 mmol,0.114 8 g)的 20 mL乙醇水溶液(3∶1,V/V),反应温度控制在50℃左右,此时溶液呈墨绿色,持续搅拌反应1 h。冷却后过滤,室温下缓慢挥发10 d析出深蓝色晶体,产物用冰无水乙醇快速洗涤。空气干燥后密封于样品管内并保存于干燥器中。
得到的配合物1易溶于二甲基亚砜(dimethyl sulpoxide,DMSO)、甲醇、乙醇等有机溶剂,但不溶于水。用元素分析仪测定配合物C、H、N原子的百分含量,按C21H20O7N4CuCl2的计算值(%):C,43.87;H,3.51;N,9.75。实验值(%):C,44.20;H,3.44;N,9.75。IR(KBr,cm-1):3 442(s,ν—OH),3 316(w,ν—NH),3 269(w,νas,—HN2),1 580(s,νas,—COO-),1 445(s,νC═N),1 317(s,νs,—COO-),,625(w,νCu—O),428(w,νCu—N)。UV-Vis(甲醇为溶剂),λ/nm(ε/(L·mol-1·cm-1)):208(28 739)、327(18 600)、627(62.65),分别归属于配体杂环的n-π*、π-π*迁移和中心铜(Ⅱ)离子的d-d跃迁。ESI-MS(甲 醇):m/z=457.8,对应于[Cu(HPBC)(LPhe)]+。电导率测定(甲醇):Λ=84 S·cm2·mol-1,表明配合物为1∶1型电解质。
1.3 配合物1晶体结构的测定
从1的母液中选择合适尺寸的单晶安装在玻璃纤维上,使用装有CCD面积检测器的Brucker Smart 1000衍射仪(石墨单色分光CuKα射线,λ=0.154 18 nm),在293(2)K下进行φ-ω扫描,以收集配合物的X射线单晶衍射强度数据。通过SAINT程序进行数据还原,并使用半经验多扫描方法(SADABS)进行了经验吸收校正[13]。所有计算应用SHELXL-2014程序包[14],采用直接法解析配合物的晶体结构,在数轮差值Fourier合成中陆续确定非氢原子的坐标,再采用全矩阵最小二乘法修正所有非氢原子坐标及其各向异性热参数。通过Fourier加氢法确定与氧相连的氢原子位置,理论加氢方法确定其他氢原子位置[7,15-16]。配合物1的晶体学数据和结构精修参数列于表1。使用SHELXLT XP升级包,并以30%的概率绘制水平热椭圆体分子结构图。
CCDC:2032899。
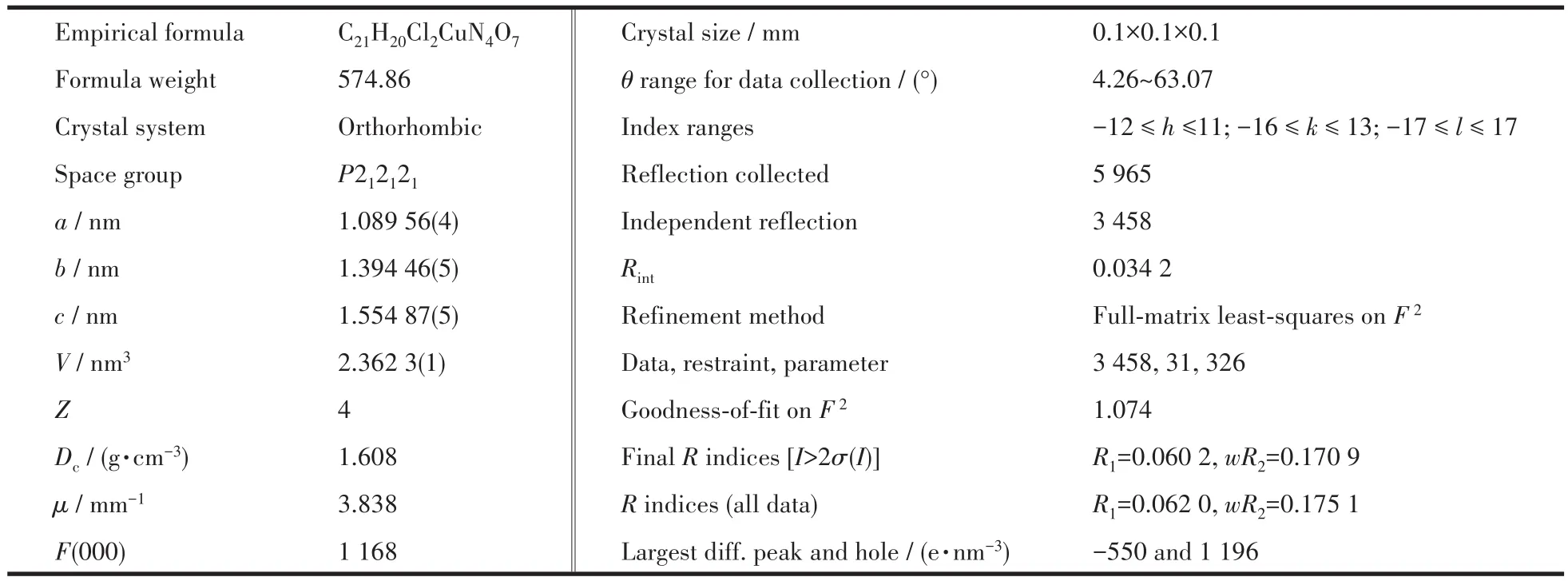
表1 配合物1的晶体学数据及结构精修参数Table 1 Crystallographic data and details of refinements for complex 1
1.4 配合物1稳定性检测
Tris-HCl/NaCl缓冲溶液(pH=7.2)的配制:称取0.06 g三羟甲基氨基甲烷(Tris)和0.29 g NaCl溶解于约95 mL去离子水中,再用稀盐酸调pH值至7.2并用去离子水定容至100 mL。后续实验的配合物溶液均用此缓冲溶液配制。
室温下,用UV-2550紫外-可见分光光度计在200~550 nm 的范围内测定了含配合物(100 μmol·L-1)的Tris-HCl/NaCl缓冲溶液(pH=7.2)在0、24和 48 h时的紫外-可见(UV-Vis)光谱。
1.5 配合物与DNA相互作用
DNA溶液的配制:称取约1 mg·mL-1的CT-DNA溶解于Tris-HCl/NaCl缓冲溶液中(约为1.5 mmol·L-1),小心振荡后放入冰箱过夜,待DNA充分溶解后测定DNA溶液在260和280 nm处的吸光度,A260/A280=1.8~1.9时,表明基本不含蛋白质,不需进一步处理。CT-DNA的浓度以碱基对的物质的量浓度(mol·L-1)计,通过测定DNA在260 nm处吸光度A260(ε=6 600 L·mol-1·cm-1),然后按下式(式 1)计算[17]:cDNA=KA260/6 600,其中K为稀释倍数。为保证实验结果的可靠性,配制好的DNA母液放置在4℃下保存,且4 d内使用。后续实验所需的DNA溶液按需用母液稀释到一定浓度。
1.5.1 紫外可见吸收滴定实验
通过保持配合物 1 的浓度恒定(50 μmol·L-1)和改变CT-DNA浓度(0~45 μmol·L-1)来进行吸收滴定实验。以Tris-HCl/NaCl缓冲溶液(pH=7.2)为扫描基线,扣除空白背景。样品池中加入3 mL不含DNA的配合物溶液,测定其在200~450 nm范围内的电子吸收光谱。为了消除由于DNA引起的吸光度变化的影响,依次滴加等量(20 μL)的 CT-DNA(1 mmol·L-1)溶液到参比池和样品池中,每次滴加DNA溶液后使其与配合物室温反应5 min,然后进行扫描。
1.5.2 荧光光谱测定
在荧光猝灭研究中,CT-DNA和EB的浓度保持恒定(分别为 10 和 8 μmol·L-1,提前混匀并静置 12 h)。具体测定过程是:往样品池中加入3 mL的EB-CT-DNA混合溶液,在240 nm·s-1、激发波长为525 nm的条件下,测定其在540~700 nm范围内的荧光发射光谱。然后,依次往EB-CT-DNA体系中滴加等体积配合物 1溶液(1 mmol·L-1,每次20 μL,体系浓度变化范围为 0~45 μmol·L-1),每次滴加后混匀,室温下孵育8 min,再测定其发射光谱。
1.5.3 粘度测定实验
用恒温水槽控制测定温度在(29.0±0.1)℃,使用乌氏粘度计测定在Tris-HCl缓冲液(pH=7.2)中不存在和存在配合物 1 时 CT-DNA(200 μmol·L-1)溶液的粘度,ccomplex/cDNA比率为0~0.30,对每个样品测量3次以确保准确性,并计算平均流动时间,同时使用EB作为阳性对照。所测溶液的相对粘度按公式η=(tt0)/t0计算[18],其中t0为缓冲溶液 Tris-HCl/NaCl的流动时间,t为含不同浓度配合物的CT-DNA溶液的流动时间。以(η/η0)1/3(η0为未加配合物时DNA溶液的比粘度)对ccomplex/cDNA作图,可以观察到配合物对DNA粘度的影响。
1.5.4 分子对接
使用Mercury软件将X射线单晶衍射的CIF文件转换为PDB格式,得到配合物1的初始结构。在进行对接计算之前,去除配合物的游离高氯酸根和配位水分子。对接使用DNA d(5'-G-dIU-TGCAAC-3')(PDB ID:454D)的晶体结构,对接前去除其中的水分子,添加极性氢原子并计算出Gasteiger电荷。整个分子对接过程在AutoDock 4.2软件上进行,使用大小为50×50×50的盒子将配体分子可能结合的区域包裹住,格点间隔为0.037 5 nm,配合物在DNA中的中心坐标为:x=29.581、y=19.637、z=70.656。采用Lamarckian遗传算法进行对接模拟,计算轮数为100次,其他参数保持默认设置。计算结果采用AutoDock 1.5.6进行分析,配合物与DNA相互作用的结果根据能量最低原则进行分析,并应用PyMol软件画出对接结果[19-20]。
1.6 配合物1的抗菌活性
抗菌实验所用菌种金黄色葡萄球菌(Staphylococcusaureus,S.aureus,G+)、李斯特菌(Listeriamonocytogenes,L.mono,G+)、大肠杆菌(Escherichiacoli,E.coli,G-)及绿脓杆菌(Pseudomonasaeruginosa,P.aeruginosa,G-)由广东省植物分子育种重点实验室提供。通过牛津杯法以抑制区为指标,分别测定了HPBC、Cu(ClO4)2和配合物的抗菌活性,每个实验设3组平行,所有设备和培养基实验前均已灭菌。
牛津杯法:用含2%DMSO的无菌水配制化合物溶液,浓度均为2 mmol·L-1。在超净工作台上,移取100 μL的菌悬液(1×106CFU·mL-1)于固体培养基上,涂布均匀并稍稍晾干,将无菌牛津杯置于含菌的固体培养基上面,然后分别将配合物、HPBC、Cu(ClO4)2及含2%DMSO的无菌水(100 μL)加入牛津杯中,置于37℃的恒温恒湿培养箱里培养24 h。用十字交叉方法测量其抑菌圈的直径,并取平均值。
1.7 配合物1的抗肿瘤活性
1.7.1 体外细胞毒性研究
采用MTT法对由中山大学实验动物中心提供的人宫颈肿瘤细胞(HeLa)、人肝癌细胞(BEL-7402)、人肺癌细胞(A549)、人胃癌细胞(SGC-7901)和人正常肝细胞(LO2)分别进行筛选,并以顺铂为阳性对照,评估配合物的体外细胞毒性。具体过程为:将实验细胞接种在96孔板(Costar,Corning Corp,New York)中,每孔细胞密度约为1×104个,并在37℃、CO2体积分数5%的培养箱中孵育,直到细胞贴壁生长至70%~80%。加入不同浓度梯度的配体及其配合物(1.57、3.13、6.25、12.5、25、50、100 μmol·L-1)作用 48 h,每组设置4个平行复孔,且设置培养液作为空白对照孔。之后,弃去培养液,各孔都加入不含血清的RPMI-1640(或 DMEM)培养基 90 μL 和MTT 溶液10 μL(5 mg·mL-1),然后继续培养 4 h,丢弃培养基,并将蓝色甲瓒固体完全溶解于DMSO中(每孔100 μL),使用多功能酶标仪测定各孔在490 nm处的光密度值,通过在对数图上绘制细胞生存率百分比与配合物浓度的关系,求出IC50值[21]。配合物抑制各种细胞增殖的活性较好,因此降低浓度梯度(0.5、1、2、3、4、5、6 μmol·L-1),进行二次筛选。
1.7.2 细胞凋亡形态
利用AO/EB染色法对凋亡细胞的形态学进行定性分析。将生长状态良好的SGC-7901细胞以每孔1×105个细胞的密度接种于12孔板中,在CO2体积分数5%的培养箱中恒温(37℃)孵育24 h后,加入不同浓度配合物(0.85、1.69 μmol·L-1)处理细胞24 h,同时设置空白组。配合物作用后弃去孔中的培养液,用PBS缓冲液清洗2次,每孔加入200 μL AO/EB混合染液(100 μg·mL-1AO,100 μg·mL-1EB,现配现用),于37℃下避光染色20 min后,弃去染液并用PBS清洗细胞。使用高内涵细胞成像系统观察细胞形态并进行拍照(整个实验应尽量避光处理)。
1.7.3 流式细胞仪检测细胞周期
将SGC-7901细胞以每孔1×106个细胞的密度接种到六孔板中,并在CO2体积分数5%的气氛中恒温(37℃)孵育24 h。除去培养基,用含有不同浓度的配合物(0.85、1.69、2.53 μmol·L-1)的培养基处理细胞24 h后,用胰蛋白酶消化细胞,收集离心后用冷PBS洗涤,再用体积分数70%的乙醇重悬细胞并在4℃下固定细胞5 h。清洗后,将20 μL的RNAse(0.2 mg·mL-1)和20 μL PI(0.02 mg·mL-1)加入细胞悬浮液中,并将混合物在37℃下孵育30 min。最后用FACS Calibur流式细胞仪分析样品,数据应用Flow-Jo软件进行分析[22]。
2 结果与讨论
2.1 配合物1的晶体结构
配合物1的部分键长和键角数据列于表2,氢键数据列于表3,其分子结构如图1所示。
单晶分析结果表明,配合物1属于正交晶系,空间群为P212121。晶体由阳离子[Cu(HPBC)(L-Phe)(H2O)]+和阴离子ClO4-构成。在配合物阳离子中,每个Cu(Ⅱ)离子和1个HPBC配体(2个N原子)、1个LPhe配体(氨基N原子和羧基O原子)及H2O(O原子)作用形成五配位的构型,这些结果与配合物的元素分析、红外光谱、紫外光谱、摩尔电导率测定及电喷雾质谱测定结果一致。另外,由于配体HPBC在溶液中易产生互变异构,结果产生的固态配合物分子中氯取代基存在着位置异构(2套无序结构在C10与C11位置各占50%)。并且发现,该配合物分子结构与文献报道的配合物[Cu(L-Val)(pzta)(H2O)]ClO4和[Cu(L-Thr)(pzta)(H2O)]ClO4相似[16]。
按照Addison/Reedijk几何构型判断标准,通过计算τ参数(τ=(β-α)/60)来初步揭示五配位配合物的变形程度[23]。计算获得配合物的τ值为0.24,表明配合物为变形四方锥构型[24]。由表2可知,配合物轴向原子到中心铜离子的距离为0.232 1 nm,大于赤道键(0.195 3~0.201 4 nm),表明配位水对中心Cu(Ⅱ)离子的配位作用较其他配体弱。此外,Cu1-N2的键长略短于Cu1-N1的键长,表明HPBC配体中咪唑N原子配位能力强于吡啶N原子,这归因于前者周围具有相对较大的电子云密度。

图1 配合物1的椭球几率为30%的分子结构图Fig.1 Molecular structure of complex 1 with thermal ellipsoids at 30% probability level
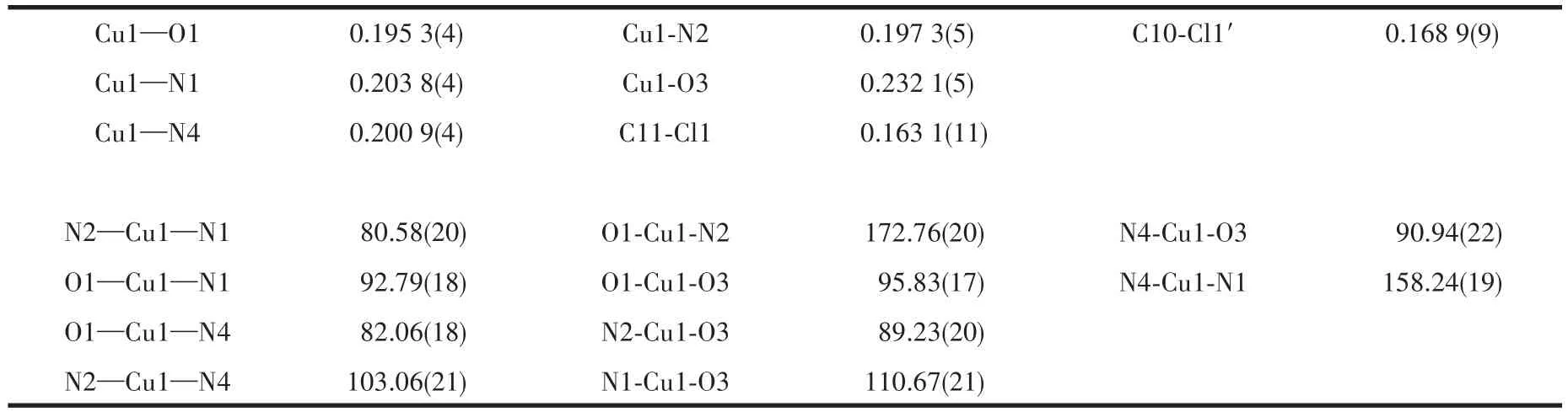
表2 配合物1的部分键长(nm)和部分键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complex 1

表3 配合物1的部分氢键参数Table 3 Selected hydrogen bond parameters of complex 1
另外,配合物晶胞堆积图(图2)揭示了配合物晶体中配位阳离子[Cu(HPBC)(L-Phe)(H2O)]+之间存在着大量的氢键(表3列出部分氢键参数)和芳环堆积作用(分子平面间质心间距离为0.343 nm,垂直距离为0.322 nm),这些作用稳定了配合物的晶体结构。

图2 配合物1的晶胞堆积图Fig.2 Unit cell packing diagram of complex 1
2.2 配合物1溶液的稳定性
图3表明,在Tris-HCl/NaCl缓冲液(pH 7.2)中,配合物的UV-Vis光谱随时间增长基本保持不变,表明配合物在溶液中能稳定存在[25-26]。因此,我们认为该配合物在后续的实验过程中是稳定的。
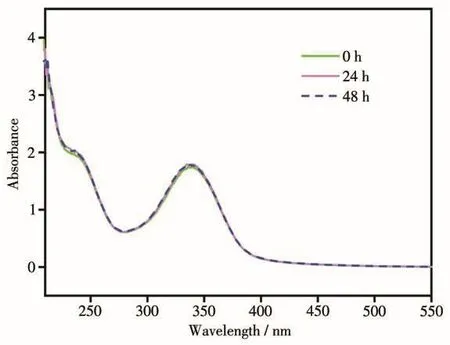
图3 配合物1在Tris-HCl/NaCl缓冲液(pH=7.2)中的紫外可见光谱Fig.3 UV-Vis spectra of complex 1 in Tris-HCl/NaCl buffer(pH=7.2)
2.3 配合物1与DNA的作用
2.3.1 电子吸收光谱
图4显示了在不存在和存在CT-DNA的情况下配合物的电子吸收光谱,虚线表示未加入CT-DNA时,配合物在Tris(pH=7.2)缓冲溶液中的紫外可见吸收光谱。吸收峰的强度随DNA浓度的增加而降低(减色率为32.65%,红移2 nm),这表明配合物可能通过插入模式与DNA结合[27]。为了评估配合物与CTDNA结合的亲和力,可以使用以下公式(式2)计算内在结合常数(Kb)[28]:cDNA/(εa-εf)=cDNA/(εb-εf)+1/[Kb(εbεf)],式中cDNA表示CT-DNA的浓度,εa、εf和εb分别表示Aobs/cCu、自由配合物的摩尔吸光系数、与CT-DNA完全结合后的配合物的摩尔吸光系数。通过cDNA/(εb-εf)对cDNA作图,得到的斜率和截距的比值即为配合物与DNA的结合常数Kb[29]。获得配合物的结合常数Kb=3.03×104L·mol-1,小于经典插入试剂EB的结合常数(1.4×106L·mol-1)[15],这可能是由于 HPBC配体的芳平面结构相对较小[30],导致配合物的插入作用较弱。

图4 配合物1(50 μmol·L-1)在不同CT-DNA浓度下的电子吸收光谱图Fig.4 Electronic absorption spectra of complex 1(50 μmol·L-1)in the absence and presence of CT-DNA
2.3.2 竞争性DNA结合实验
配合物对EB-CT-DNA体系的荧光猝灭光谱如图5所示。显然,随着体系中配合物浓度不断增加,荧光强度不断减弱,发生了猝灭现象(荧光淬灭率为24.35%),表明配合物挤出了DNA碱基对间的EB,从而形成新的配合物-CT-DNA体系,即配合物与CT-DNA发生了插入结合作用。根据经典的Stern-Volmer公式(式3)可定量计算出荧光猝灭常数KSV[31]:F0/F=1+KSVccomplex,其中,F0和F分别表示未加入和加入配合物时EB-CT-DNA体系的荧光强度,ccomplex表示配合物的浓度。以F0/F对ccomplex作图获得直线的斜率即为KSV值。获得配合物对EB-DNA体系的荧光猝灭常数为5.365×103L·mol-1,稍小于文献中结构相似的铜配合物[32-33],即揭示了该配合物对DNA的插入结合作用较弱,与上述电子吸收光谱方法测定结果一致。
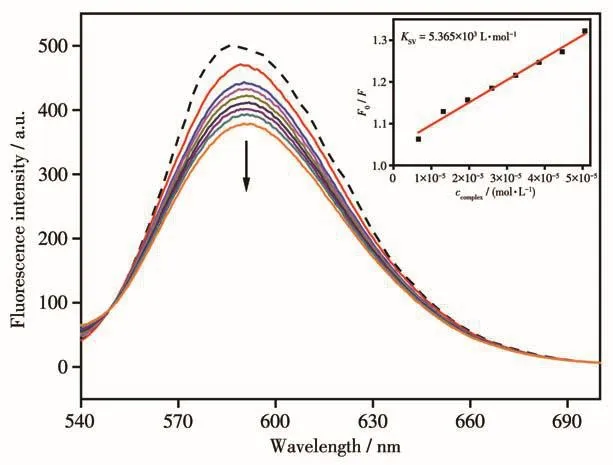
图5 配合物1对EB-CT-DNA体系荧光强度的影响Fig.5 Emission spectra of EB-CT-DNA in the absence and presence of complex 1
2.3.3 粘度测定
DNA粘度对其链长度变化比较敏感,是检测溶液状态下配合物与DNA相互作用模式的最有效的手段[34]。图6表明,随着EB浓度的增加,DNA溶液的相对粘度大幅度增加;而随着配合物的浓度不断增加,CT-DNA溶液的相对粘度有所升高但趋势并不明显,结合以上光谱方法实验结果,推测配合物与DNA之间具有弱的插入作用。
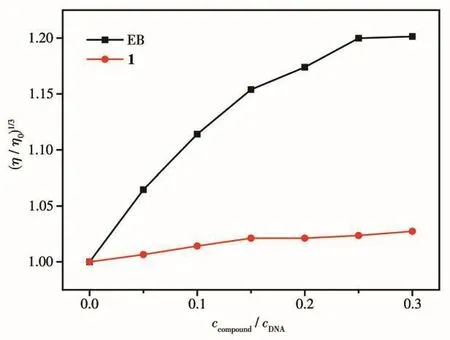
图6 不同浓度的配合物1或EB对CT-DNA溶液相对粘度的影响Fig.6 Effect of increasing concentration of complex 1 or EB on relative viscosity of CT-DNA
2.3.4 分子对接
将合成的配合物与DNA(PDB ID:454D)进行了分子对接模拟,以揭示配合物与DNA之间的相互作用方式。本工作对配合物与DNA分子对接100次的结果进行能量分析,并根据能量最低原则,选取能量最低时所对应的结合状态如图7所示。结果表明,配合物与DNA之间的结合能为-35.10 kJ·mol-1,配合物通过其疏水性的芳杂环配体HPBC插入到了DNA中心位置的GC/GC碱基的空腔之间(配体与碱基间距离为0.36 nm),表明配合物与DNA之间存在插入作用,印证了上述实验得到的结论。同时,配合物与DNA之间形成了一个氢键:Complex:H4A…454D:DC5-5:O5'(0.20 nm),这种作用进一步增强了配合物与DNA的作用。
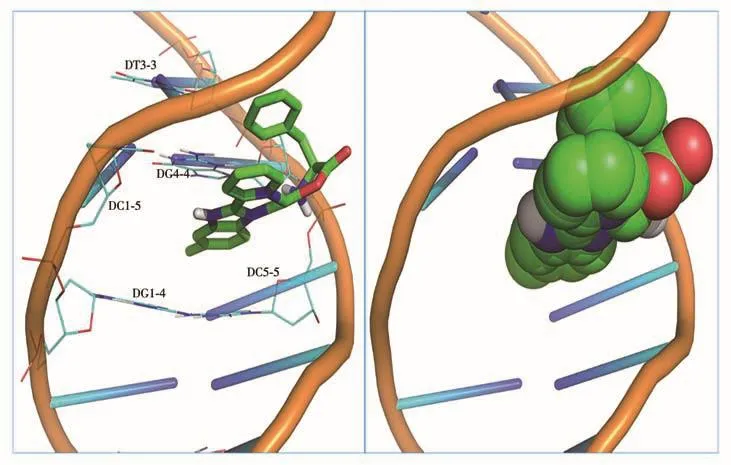
图7 配合物1与DNA(PDB ID:454D)作用的分子对接模型图Fig.7 Molecular docking of DNA(PDB ID:454D)-complex 1 interactions
2.4 抗菌活性
用含2%DMSO的无菌水作为阴性对照,测得HPBC、Cu(ClO4)2和配合物 1 对S.aureus(G+)、L.mono(G+)、E.coli(G-)及P.aeruginosa(G-)等 4种细菌的抑菌圈直径如表4所示。
结果表明,含2%DMSO的无菌水及配体HPBC无抗菌活性,且在相同测试浓度和同一菌种的条件下,配合物的抑菌圈直径明显大于Cu(ClO4)2,表明配合物有相对较强的抑菌活性,这可能与金属铜(Ⅱ)离子与配体HPBC间的螯合作用有关。中心铜(Ⅱ)离子与HPBC配位后,其正电荷部分转移到配体上而产生了离域效应,进而增加了脂溶性,更容易穿透细菌细胞膜[35]进而抑制细菌的生长。另外,配合物对2种革兰氏阳性菌(S.aureus、L.mono)都具有较好的抑菌活性,但对革兰氏阴性菌具有选择性,能抑制E.coli(G-)的增长,但对P.aeruginosa(G-)的抑制活性并不明显,这可能归因于革兰氏阳性菌与阴性菌的细胞壁组成成分及结构明显不同[36]:革兰氏阳性菌细胞壁较厚,但结构较简单,含大量的肽聚糖及磷壁酸;而革兰氏阴性菌细胞壁较薄,但结构较复杂,分为外壁层和内壁层,主要含有脂多糖和脂蛋白。这导致这2类细菌对某些药物的敏感性有很大差异[37-38]。

表4 HPBC、Cu(ClO4)2及配合物1对不同菌种的抑制活性Table 4 Inhibitory activity of HPBC,Cu(ClO4)2and complex 1 against different microorganisms
2.5 抗肿瘤活性
2.5.1 体外细胞毒性
采用标准MTT比色法测得配体HPBC、配合物1和顺铂对各种细胞的毒性作用如图8、表5所示。
图8及表5表明,与配体相比,配合物1对4种癌细胞株的抑制活性显著增强,具有广谱的细胞毒性,且抑制活性强于传统抗癌药物顺铂,但对人正常肝细胞(LO2)的选择性仍然较低。与结构相似配合物[Cu(HPBM)(L-Phe)(H2O)]ClO4(IC50值为 5.7~8.3 μmol·L-1)[39]相比,1的细胞毒性进一步增强,这可能主要归因于HPBC中卤素(—Cl)取代基与芳香苯并咪唑环形成p-π共轭体系,导致环上π电子云密度加大从而可增强配合物的脂溶性、提高配合物与细胞的亲和力,进而增强其细胞毒性作用[40-41]。此外,配合物对不同肿瘤细胞株的抑制活性大小为SGC-7901>A549>HeLa>BEL-7402,即对胃癌细胞的抑制效果最为显著(IC50=1.69 μmol·L-1),故选用SGC-7901细胞进行后续机制探究实验。
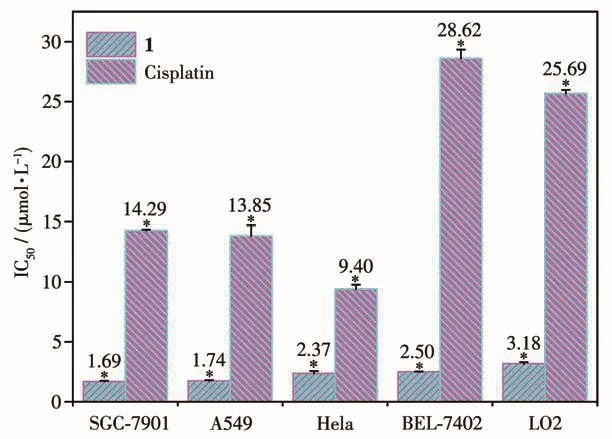
图8 配合物1与顺铂的细胞毒性IC50值柱状图Fig.8 Histogram of IC50values for the cytotoxicity of complex 1 and cisplatin

表5 配体HPBC及配合物1的体外细胞毒性Table 5 In vitro cytotoxicity of HPBC and complex 1
2.5.2 AO/EB染色
为观察配合物1作用后细胞的形态学变化,采用AO/EB双染法进行分析。分别用0.85和1.69 μmol·L-1的配合物1作用于SGC-7901细胞,经AO/EB染色后拍照,结果如图9所示。从图中可以观察到,正常SGC-7901细胞(a)的细胞核形态正常且细胞质分布均匀,呈现均匀的浅绿色荧光。当以不同浓度配合物处理细胞24 h后,发现细胞变圆,细胞质分布不均匀,细胞核固缩,出现了亮绿色的荧光,并且随着配合物浓度的增大细胞固缩更加明显,表明主题配合物可以诱导细胞发生凋亡并存在浓度依赖性。
2.5.3 细胞周期阻滞

图9 不同浓度配合物1作用于SGC-7901细胞24 h的凋亡形态分析Fig.9 Apoptosis morphology analysis of SGC-7901 cells exposed to different concentrations of complex 1 for 24 h

图10 不同浓度配合物1对SGC-7901细胞周期的影响Fig.10 Effect of different concentrations of complex 1 on SGC-7901 cell cycle
经配合物1作用后,使用流式细胞仪对经PI染色的细胞进行分析,细胞周期分布如图10所示。结果表明,在空白组细胞中,处于G2/M期的细胞比例为22.39%,经不同浓度配合物1作用24 h后,G2/M期的细胞比例分别为30.45%(0.85 μmol·L-1)、33.19%(1.69 μmol·L-1)和 48.81%(2.53 μmol·L-1),处于该期的细胞比例分别增加了8.06%、10.80%和22.42%。与此相对应的是,处于G0/G1期的细胞相应减少,揭示了配合物1可将细胞周期阻滞在G2/M期。
3 结论
合成、表征了新的三元铜(Ⅱ)配合物[Cu(HPBC)(L-Phe)(H2O)]ClO4,该配合物具有五配位的变形四方锥构型。多种实验结果表明,配合物通过插入方式与DNA相互作用,并且分子对接结果进一步揭示了配合物通过芳杂环配体插入到GC/GC残基区域的空腔中。生物实验结果表明该配合物对实验测试的部分细菌(S.aureus、L.mono、E.coli)和所有肿瘤细胞表现出良好的抗微生物活性和细胞毒性作用。其中,配合物对SGC-7901细胞抑制活性最强且远大于顺铂,同时揭示了配合物通过DNA结合的途径损伤DNA,将细胞周期阻滞在G2/M期并进一步诱导SGC-7901细胞凋亡。研究结果对设计、合成新的抗菌、抗肿瘤活性金属配合物药物具有一定意义。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13 08:59:28
中成药(2021年5期)2021-07-21 08:38:32
上海包装(2019年2期)2019-05-20 09:10:52
中央民族大学学报(自然科学版)(2018年3期)2018-11-09 01:16:32
中成药(2018年2期)2018-05-09 07:19:49
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
微生物与感染(2015年1期)2015-02-28 17:42:37
无机化学学报(2014年6期)2014-02-28 17:32:06
无机化学学报(2014年5期)2014-02-28 17:31:42

- 无机化学学报的其它文章
- 《无机化学学报》投稿须知(NOTICE TO AUTHORS)
- 三维α-MnO2@Co3O4异质材料的制备、表征及其催化性能
- Aluminum Amine Compound Protected by β-Diketiminate Ligand:Preparation and Enhanced Performance as Catalyst for Ring-Opening Polymerization of ε-Caprolactone
- Preparation,Structures and Thermal Stabilities of Four Transition Metal Complexes Constructed by 3,7-Di(3-pyridyl)-1,5-dioxa-3,7-diazacyclooctane Bipyridine Ligand
- 基于1D/0D有序复合SnO2纳米晶的钙钛矿太阳能电池
- 巯基/羧基修饰硅藻土及其对Pb(Ⅱ)、Cd(Ⅱ)的吸附性能