
DNA酶1样蛋白3基因及编码蛋白在系统性红斑狼疮中的研究进展*
2021-02-23 02:59张乃丹翟建昭综述武永康审校
国际检验医学杂志 2021年3期
张乃丹,翟建昭,张 苹 综述,武永康,2△ 审校
四川大学华西医院:1.实验医学科;2.门诊部,四川成都 610041
系统性红斑狼疮(SLE)的特征是产生针对核抗原,如RNA和DNA的一组自身抗体,其中,B细胞对DNA和(或)染色质耐受性丧失是SLE发病的重要机制。自身反应性B细胞受抗原刺激后增殖活化,参与表面抗原CD4+T细胞分化并激活固有免疫系统,包括浆细胞样树突状细胞和分泌过量的Ⅰ型干扰素(IFN-Ⅰ)[1]。凋亡细胞DNA降解失活、嗜中性粒细胞胞外捕获物和氧化线粒体DNA等先天免疫反应加剧B细胞活化,进一步诱导了SLE的发生与发展。通过对DNA酶1样蛋白3(DNASE1L3)基因缺失的小鼠模型及儿童SLE患者家系病例的研究,结果显示,DNASE1L3基因纯合突变DNASE1L3:c.643delT与血清抗双链DNA抗体相关,并与狼疮性肾炎的高发病率相关[2-3]。由于缺乏性激素,儿童SLE发病可能提示遗传因素占据主导地位,这提供了SLE致病基因遗传多态性研究的新思路。本文将从DNASE1L3基因及编码蛋白Dnase1L3的特征、在SLE发病机制中的作用,以及DNASE1L3遗传学在SLE中的研究进展等方面进行综述,为SLE遗传因素与免疫细胞的相关研究提供参考依据。
1 DNASE1L3基因及编码蛋白Dnase1L3的特征
1.1DNASE1L3基因和编码蛋白Dnase1L3的结构特征 DNASE1L3基因位于人类染色体3p14.3,含有8个外显子。该基因编码脱氧核糖核酸酶1(Dnase1)家族中的蛋白3(Dnase1L3),后者由275个氨基酸构成[4]。Dnase1L3的C端是螺旋结构(CTH),并且含有核定位序列,核定位序列可加速细胞外Dnase1L3进入凋亡微粒细胞膜。研究显示,Dnase1L3的C端结构在其发挥消化细胞外微粒相关染色质和清除血清抗双链DNA抗体等生物学效应方面显示出重要作用[5-6]。见图1。
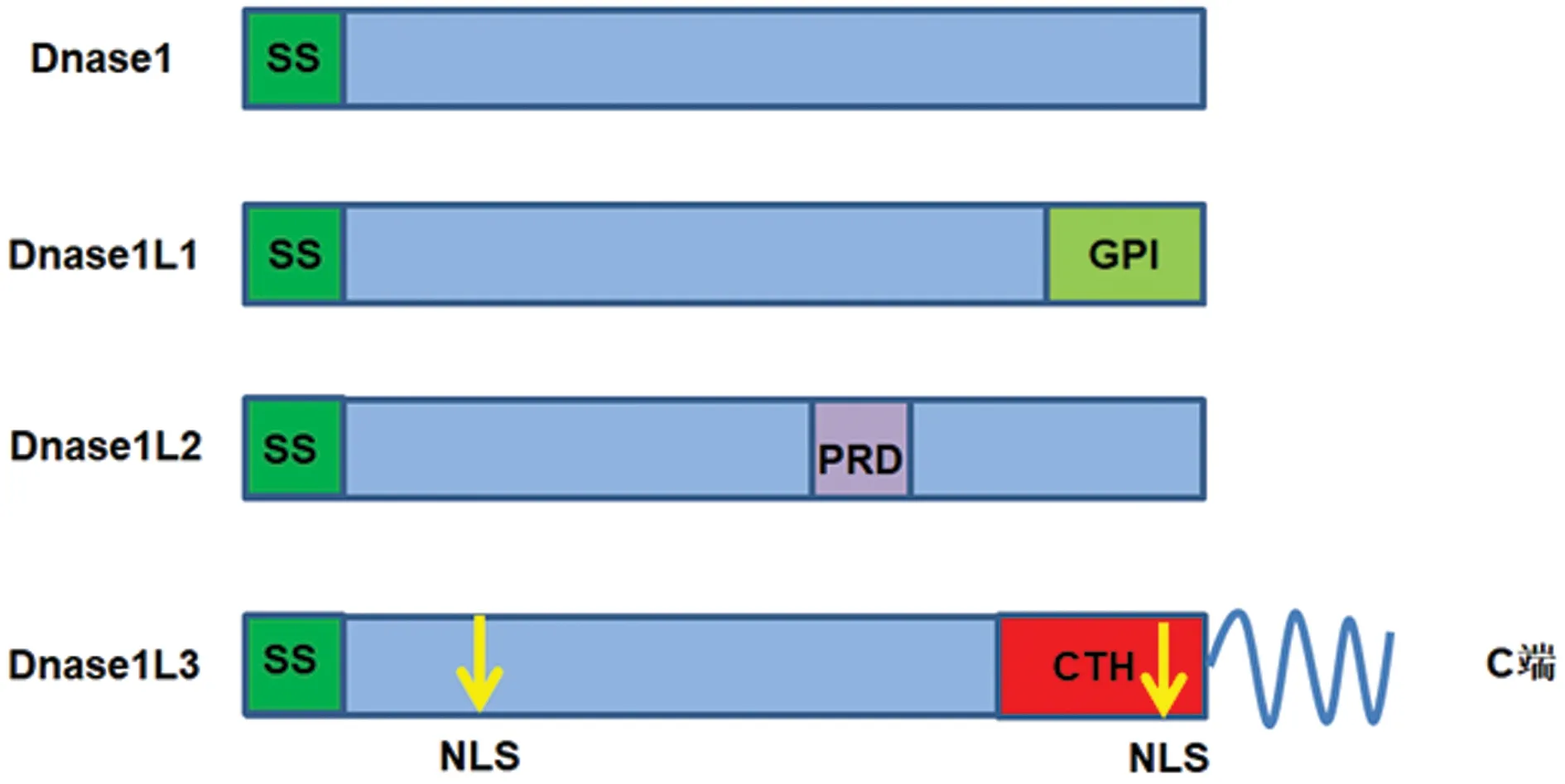
注:SS表示信号序列;GPI表示糖磷脂酰肌醇锚定序列;PRD表示富脯氨酸域;NLS表示核定位序列;CTH表示C端螺旋。
1.2DNASE1L3基因和编码蛋白Dnase1L3在自身免疫性疾病中的作用
1.2.1Dnase1L3发挥细胞外作用与自身免性疫病 Dnase1L3发挥细胞外和细胞内作用共同预防自身免性疫病的发生。细胞外作用包括Dnase1L3与Dnase1形成阻断质粒转染的嵌合蛋白,以及通过免疫细胞靶向DNA-蛋白质复合物,降解中性粒细胞胞外诱捕网(NETs)限制炎症发作期间的组织损伤[7]。Dnase1L3通过清除SLE患者体内来自NETs和凋亡细胞的核酸,参与核小体DNA碎片化,有效避免了滤泡树突状细胞激活并呈递给自身反应性B细胞,抑制了抗双链DNA抗体产生[6]。研究显示,Dnase1L3缺陷与SLE患者体内NETs降解延迟和血管损伤密切相关,导致狼疮性肾炎病情加重[8]。
1.2.2Dnase1L3发挥细胞内作用与自身免疫性疾病 Dnase1L3在细胞内主要参与抑制环状GMP-AMP合成酶(cGAS)-干扰素激活蛋白(STING)途径。SLE患者细胞核或线粒体DNA释放到细胞质,与cGAS作用生成环鸟腺苷酸(cGAMP)。cGAMP结合并激活STING,STING招募TANK结合激酶(TBK1)并转位至核内体,激活干扰素调节因子3(IRF-3),介导IFN-α/β产生[9]。Dnase1L3还通过调节高迁移率族蛋白B1(HMGB1)释放,介导自身反应性B细胞增殖和活化[10]。活化B细胞产生HMGB1/DNA免疫复合物的累积效应促使炎症细胞因子分泌增加,最终导致SLE的发生[11]。动物模型研究显示,DNASE1L3基因缺陷导致小鼠巨噬细胞分泌大量白细胞介素(IL)-1β[12]。对缺乏DNASE1L3基因小鼠进行IFN-β注射,通过激活炎性小体,小鼠迅速发展出与SLE相关的多种免疫并发症[5]。上述研究提示DNASE1L3基因和编码蛋白Dnase1L3通过抑制cGAS-STING途径调节B细胞,抑制促炎因子分泌,预防自身免疫性疾病的发生。
2 DNASE1L3基因和编码蛋白Dnase1L3在SLE发病机制中的作用
2.1DNASE1L3基因和编码蛋白Dnase1L3与B细胞异常 Dnase1L3在清除凋亡细胞DNA方面发挥重要作用,凋亡细胞DNA作为自身抗原,一方面通过TLR3、7、9激活B细胞,促进浆细胞激活和抗双链DNA抗体的产生;另一方面,经凋亡细胞DNA免疫后的B细胞可以刺激CD4+T细胞分化增殖,激活更多B细胞。动物模型显示,肝脏组织中Dnase1L3清除循环微粒DNA,阻止DC细胞和Kupffer细胞通过toll样受体(TLRs)和受体损伤相关分子模式(DAMP)介导CD4+T细胞分化[13-14]。DNASE1L3基因缺失的C57BL/6小鼠在SLE疾病早期出现了滤泡辅助性T细胞(Tfh)活化及脾脏中浆细胞数量增加[15-16]。Tfh活化产生CD40L、IL-21加速B细胞激活及自身抗体产生。研究表明,CD40与CD40L结合招募肿瘤坏死因子受体相关因子(TRAFs)与肿瘤坏死因子受体相关细胞适配器蛋白、TLRs和IL-1受体,激活并引发丝/苏氨酸蛋白激酶(MAPK),磷酸肌肽3-激酶(PI3K)和核转录因子κB(NF-κB)途径及促炎因子表达,导致B细胞异常活化而产生自身抗体[17]。见图2。
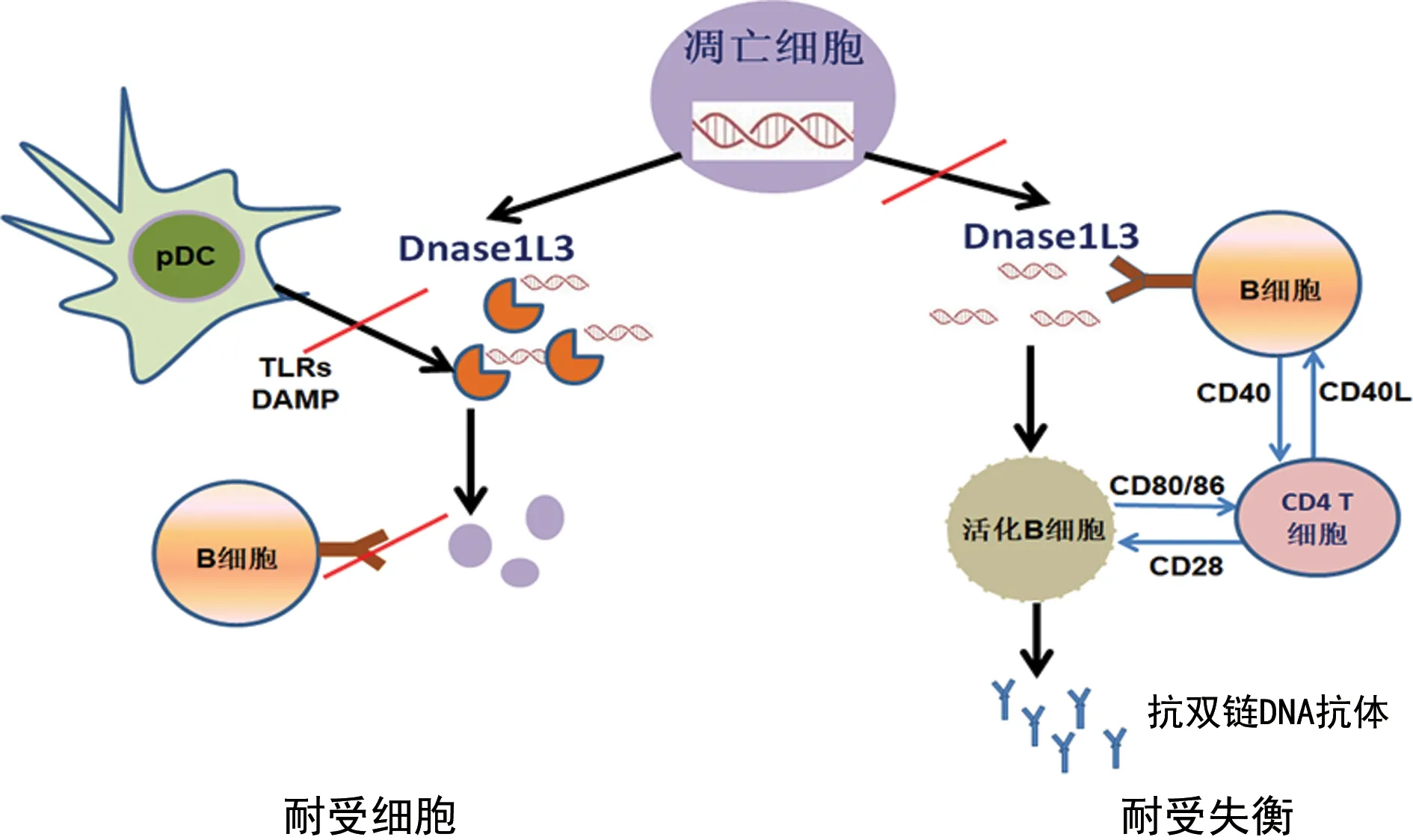
图2 Dnase1L3与SLE患者B细胞异常的作用示意图
2.2DNASE1L3基因和编码蛋白Dnase1L3与细胞因子
2.2.1IL IL-4是由Ⅱ型辅助T细胞(Th2)产生,诱导Th2细胞因子转换,有效抑制促炎因子IL-6、IL-17、IL-12、肿瘤坏死因子α的表达。研究显示,IL-4是髓样细胞和单核巨噬细胞中DNase1L3表达的重要调控因子[18]。IL-4诱导单核细胞衍生DC中DNase1L3 mRNA表达,通过细胞外调节蛋白激酶/PI3K信号通路促进DNase1L3分泌到细胞外,降解凋亡细胞DNA,在组织稳态和预防自身免疫性疾病中发挥了重要作用[3,19]。
2.2.2IFN-Ⅰ IFN-Ⅰ包括IFN-α和IFN-β,IFN-Ⅰ信号通路在人类和小鼠SLE中的致病作用已经得到证实[20]。关于IFN-Ⅰ信号通路在SLE中的致病作用的研究集中在产生IFN-Ⅰ的细胞类型和IFN-Ⅰ信号转导的靶细胞上[21]。对Dnase1L3缺陷小鼠研究显示,Dnase1L3缺陷导致的DNA降解失活和抗原游离DNA暴露导致TLRs依赖的pDC活化,产生IFN-α。IFN-α升高通过外周T滤泡辅助细胞(ExFO-Th)途径促进抗双链DNA抗体形成细胞持续外分化,从而驱动SLE的病理效应[15]。由于pDC在体内产生的IFN-Ⅰ难以检测,DNASE1L3基因遗传学证据和pDC体外IFN-Ⅰ驱动模型研究将会为治疗SLE提供新的治疗策略。
3 DNASE1L3基因遗传学在SLE中的研究进展
DNASE1L3基因多态性与自身免疫性疾病易感性相关,如系统性红斑狼疮、类风湿关节炎、强直性脊柱炎和血管炎等,见图3。截至2020年5月,关于DNASE1L3基因多态性与SLE相关研究的文献数量明显多于其余各类疾病,提示充分研究DNASE1L3基因多态性与SLE发病机制的关系有望为治疗SLE带来新的靶点。
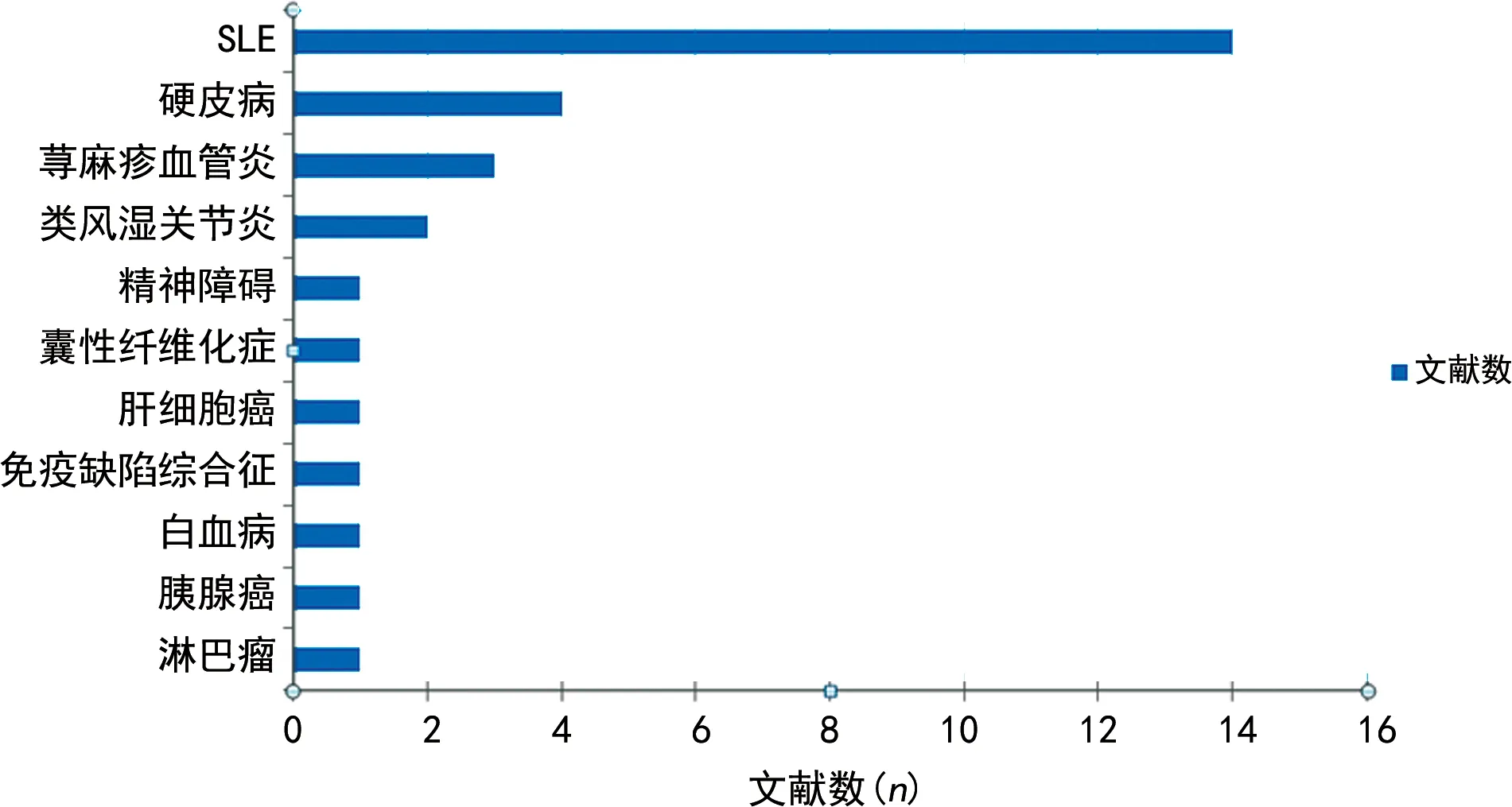
注:横轴表示PubMed检索到的文献数,纵轴表示与DNASE1L3基因相关的疾病种类。
3.1DNASE1L3基因多态性与SLE 研究显示DNASE1L3基因的单核苷酸多态性(SNP)位点与SLE发病具有一定的相关性,特别是在DNASE1L3基因缺失的小鼠模型和SLE家系中[15,22]。大部分疾病相关SNP位点位于 DNASE1L3基因编码区上、下游或附近内含子区域,提示这些基因位点突变可能通过与相邻外显子作用,干扰该基因的调控进而影响Dnase1L3蛋白的表达。关于DNASE1L3基因甲基化和组蛋白修饰的研究尚未明确,提示DNASE1L3基因表观遗传修饰功能还需进一步的研究。
有研究采用全基因组关联分析的方法对沙特阿拉伯地区儿童SLE的7个家系进行DNASE1L3基因测序,研究显示在DNASE1L3 (NM_004944.2)中发现c.289_290delAC (rs751206379)纯合突变与SLE疾病易感性密切相关,该突变属常染色体隐性突变,影响了Dnase1L3酶活性功能;自噬酶分析显示rs751206379无效突变与儿童狼疮性肾炎相关,患者临床血清学表现为抗双链DNA抗体阳性、低补体(C)3和C4;该研究指出,DNASE1L3基因多态性可能影响PXK基因编码蛋白的表达[23]。PXK基因位于3p14.3,并且DNASE1L3基因是PXK上游140 kb的唯一基因[24]。该研究为SLE发病机制的研究提供了新视角,即导致细胞内双链DNA 或RNA传感通路异常调节和激活的单基因突变可能导致异常IFN-Ⅰ产生,随后促进自身免疫炎性反应和自身抗体的产生。
2013年OZÇAKAR等[25]采用Affymetrix 5.0芯片进行全基因组SNP基因分型后证实,来自土耳其地区的儿童SLE家系中3例患者检出c.289_290delAC纯合突变。值得注意的是,3例患者在确诊为SLE之前出现低补体血症荨麻疹性血管炎的临床表现[26]。SNP基因分型鉴定了新的突变位点c.320+4delAGTA(rs766912371),该突变位点位于内含子3,导致多肽序列中第78位天冬酰胺到第107位赖氨酸发生了缺失突变,Dnase活性分析显示rs766912371突变与Dnase1L3活性降低有关[25]。随后,UEKI等[27]采用限制性片段长度多态性聚合酶链反应(PCR-RFLP)技术在3个种族(亚洲人、非洲人和高加索人)共计1 752例SLE患者中检测并计算了DNASE1L3基因每个SNP的小等位基因频率和杂合度,结果鉴定出DNASE1L3基因的5个功能突变位点:rs145888358、rs146805633、rs141477807、rs1472 19402和rs148314913,见表1。上述位点突变与Dnase1L3活性显著降低有关[29]。由于这些位点变异编码的氨基酸位于功能关键氨基酸残基附近,关于DNASE1L3基因与SLE靶基因的相互关系需要进一步研究证实。
3.2微小RNA(miRNA)调节DNASE1L3基因表达调控 miRNA是具有调控功能的非编码RNA,通过细胞核内转录剪切、输出到细胞质并最终成为成熟miRNA。成熟miRNA与RNA诱导沉默复合物结合,诱导靶mRNA剪切或阻止其翻译对靶基因进行表观修饰,进一步影响靶基因表达和信号通路传导,与DNASE1L3基因有关的miRNA研究见表2。研究显示,miR-1184参与了通过转化生长因子(TGF)-β途径控制DNA双链断裂的修复过程[30]。关于miR-142-3p/5p的研究显示,B细胞淋巴瘤6蛋白通过上调CD4+T细胞中的H3K27me3和下调H3K9/K14ac抑制miR-142-3p/5p的表达,导致T细胞过度活跃和B细胞过度刺激,促进SLE的发生[31]。尿液中miRNA的研究显示,miR-335和miR-146a在狼疮性肾炎患者中表达上调,并且miR-146a具有区分活动性狼疮性肾炎的效能[32]。
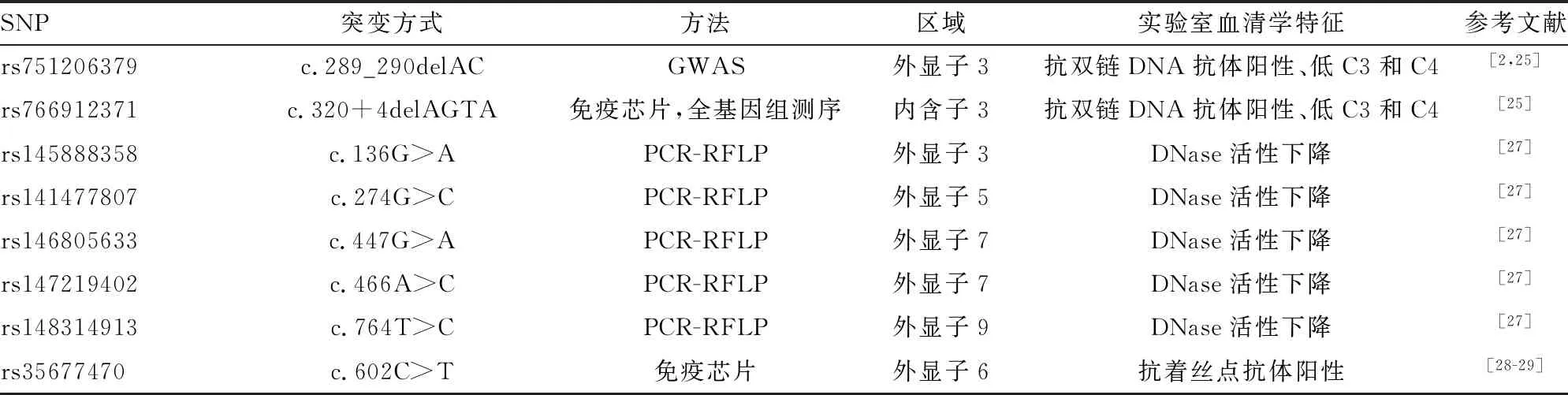
表1 DNASE1L3基因中SNP的研究位点及功能评价
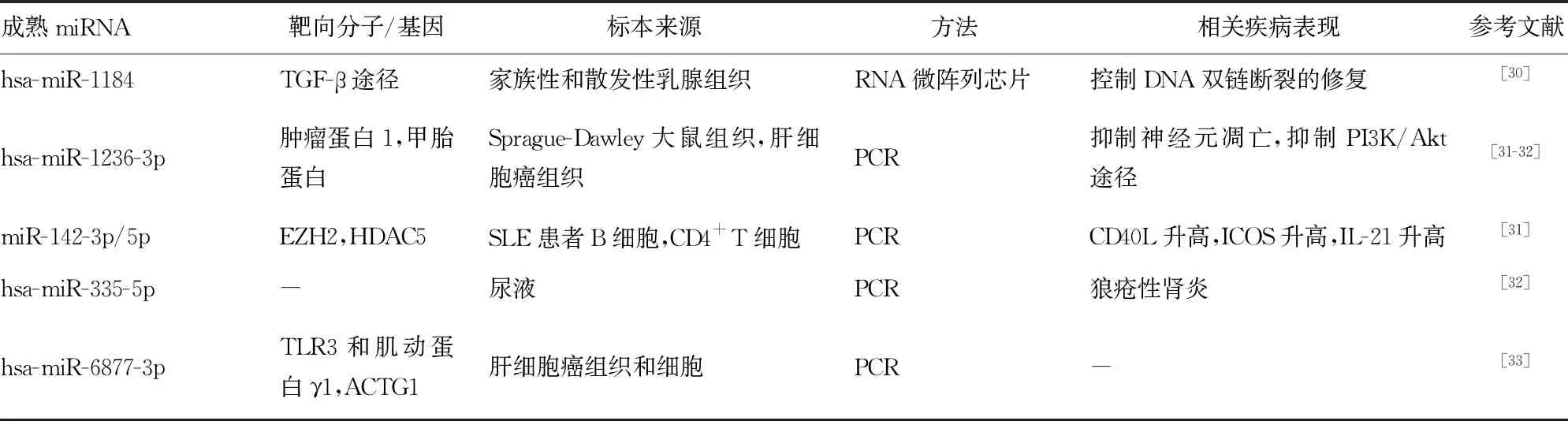
表2 DNASE1L3基因中miRNA的研究位点相关研究信息
3.3Dnase1L3与细胞游离DNA 细胞游离DNA是循环游离DNA或者释放到血浆中的降解DNA片段,主要来源于凋亡和坏死细胞、移植物或胎儿游离DNA及自主激活释放的DNA。目前已经广泛用于产前无创检测、器官移植状态监控及无创肿瘤DNA研究等方面[34]。Dnase1L3作为清除凋亡细胞的重要酶类,Dnase1L3缺陷小鼠模型显示,Dnase1L3作为内切酶,首先在对乙酰氨基酚诱导的坏死肝细胞中生成细胞游离DNA,胱天蛋白酶激活的Dnase和Dnase1L3协同产生细胞游离DNA用于抗Fas介导的肝细胞凋亡[35]。另有研究显示,在Dnase1L3缺陷小鼠中出现血浆细胞游离DNA片段的畸变。这种畸变包括120 bp以下增加的短DNA分子及血浆DNA末端基序的频率异常,且畸变频率与血清中抗DNA抗体水平呈正相关[36]。表明Dnase1L3作为细胞游离DNA的调节剂,可能对该酶阻止自身免疫性疾病的发生具有重要意义。
4 小 结
DNASE1L3基因突变及编码蛋白Dnase1L3活性丧失可导致凋亡微粒中染色质和DNA清除障碍,Dnase1L3是促进细胞凋亡相关斑点样蛋白所必需的酶,Dnase1L3通过靶向caspase和募集域介导细胞凋亡和后续炎性因子释放[37]。DNASE1L3基因突变和编码蛋白Dnase1L3清除凋亡DNA功能异常,导致Tfh细胞产生的IL-4、IL-17、IL-21与活化B细胞相互作用,最终导致致病性自身抗体的产生。
近年来多项研究分别从动物模型、细胞实验和人体标本的角度来探索DNASE1L3基因及编码蛋白Dnase1L3在SLE发生、发展中的作用,但其具体机制及与SLE靶基因之间的相互关系尚未完全阐明。通过回顾不同地区、种族之间SLE家系及动物模型的研究显示,DNASE1L3基因突变可能预示着SLE在不同年龄阶段的致病机制具有较大差异,如儿童SLE与成人SLE。虽然单基因和孟德尔遗传性狼疮较为少见,但相关研究提示对SLE易感基因的研究应该充分考虑种族和地区的差异,应对患者进行年龄、性别、不同疾病表型等更为详细的分层。同时,表观遗传修饰在SLE相关致病基因中的调控机制也是值得关注的问题。DNASE1L3基因的位点突变导致凋亡DNA和染色质清除障碍,异常调控的表观遗传效应会对免疫细胞功能产生巨大影响。随着分子诊断技术的不断发展和对DNASE1L3基因及编码蛋白Dnase1L3研究的不断深入,DNASE1L3基因及编码蛋白Dnase1L3在SLE疾病发生、进展及预后中的作用将有助于SLE靶向治疗的应用。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
汉字汉语研究(2020年2期)2020-08-13
电子制作(2019年22期)2020-01-14
生物学通报(2019年3期)2019-02-17
疯狂英语·新读写(2018年3期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
中央民族大学学报(自然科学版)(2015年1期)2015-06-11
