
以发作性运动障碍和感觉障碍为主要表现的成人苯丙酮尿症1例报告
2021-02-23 09:31张国荣王玉忠冯勋刚
中风与神经疾病杂志 2021年1期
汪 燕, 刘 晨, 张国荣, 王玉忠, 冯勋刚
苯丙酮尿症(phenylketonuria,PKU)是一种常见的遗传代谢性疾病,患者由于缺乏苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)或辅酶四氢生物蝶呤,导致体内酪氨酸浓度降低和苯丙氨酸沉积,其常见的表现为鼠臭味尿液、毛发黄、智力低下、癫痫发作等。PKU多见于儿童患者,成人PKU报道少见且临床特征不典型,这给成人PKU的诊断和治疗造成很大的困难[1,2]。本文报道1例表现为发作性运动障碍和感觉障碍的成人PKU,并结合相关文献探讨该病的发病机制及临床特点,旨在提高神经内科医生对成人PKU的认识。
1 临床资料
患者,男,18岁,因“左上肢抖动半年,麻木1月余”入住我科。既往无心脑血管病、癫痫病史,无高蛋白摄入史。家中独子,父母均体健。患者半年前无明显诱因出现左上肢抖动,多于持物时发作,每次持续约10 min,休息后可缓解,发作频率逐渐增加,未诊治。1月余前出现左上肢麻木,自诉酸痛。入院查体:体温36.9℃,脉搏89次/min,呼吸20次/min,血压123/69 mmHg,头颅及毛发未见异常,心肺腹无异常。神志清,精神可,言语流利,定向力及记忆力正常,智力发育正常,颅神经查体无阳性体征,四肢肌力、肌张力正常,无肌肉萎缩,四肢腱反射正常,左上肢痛觉减低,深感觉、共济实验无异常,双侧巴宾斯基征阴性,脑膜刺激征阴性。辅助检查:血常规、电解质、肝肾功能、甲状腺功能、风湿免疫相关检查均正常,血清同型半胱氨酸39.1 μmol/L。颅脑MR平扫:双侧侧脑室周围可见条片状T2WI、DWI高信号影,考虑急性脱髓鞘改变(见图1),行钆喷酸葡甲胺增强扫描后未见病灶强化;颅脑MRA、MRS及颈椎MRI平扫均未见异常;四肢神经传导速度示四肢神经传导正常,体感诱发电位提示中枢病变。初步诊断:颅内异常信号原因待查,脱髓鞘脑病?为进一步明确是否为免疫介导的中枢神经系统脱髓鞘疾病,完善腰穿检查提示:脑压109 mmH2O;脑脊液蛋白0.45 g/L,脑脊液免疫球蛋白A 6.61mg/L,较正常值偏高;脑脊液常规、真菌涂片、一般细菌涂片、抗酸杆菌涂片及墨汁染色均正常;脑脊液-IgG寡克隆区带阴性。患者为青年男性,血清同型半胱氨酸升高,不能排除遗传代谢性疾病,予以完善血氨基酸串联质谱筛查提示:苯丙氨酸、苯丙氨酸/酪氨酸浓度比值增高,谷氨酰胺、谷氨酰胺/瓜氨酸、蛋氨酸/苯丙氨酸、缬氨酸/苯丙氨酸降低;尿串联质谱筛查发现苯丙氨酸和苯丙氨酸代谢产物中苯乙酸、扁桃酸盐及2-羟基苯乙酸的尿排泄量超出正常浓度范围,其他代谢产物无异常。基因检测显示PAH基因发现外显子7中c.722G>A、c.728G>A的杂合核苷酸变异,均可导致其编码的PAH功能受到影响;其父亲的第7外显子也发现c.728G>A的杂合变异,但是第722号核苷酸未发现变异,其母亲因个人原因未能行基因检测。结合患者病史、体征及辅助检查,患者诊断明确:(1)苯丙酮尿症;(2)高同型半胱氨酸血症。嘱患者低苯丙氨酸饮食,给予维生素B12、叶酸降低同型半胱氨酸治疗并出院随访。半年后患者门诊复诊,未再出现肢体抖动及感觉异常,复查血清同型半胱氨酸降至正常。
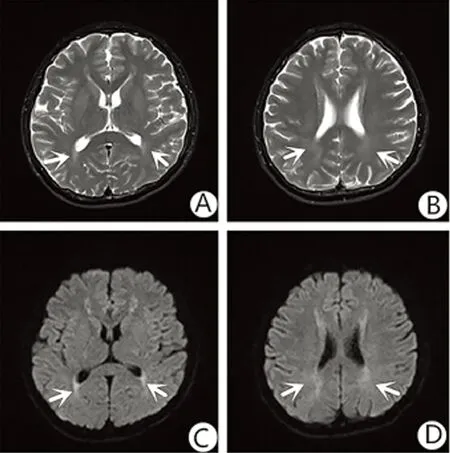
图1 颅脑MR平扫显示双侧侧脑室周围见条片状T2WI稍高信号影(A、B)、DWI呈高信号(C、D),考虑急性脱髓鞘改变(箭头所示)
2 讨 论
PKU是一种常染色体隐性遗传病,最常见的原因是编码PAH的基因发生突变,该基因定位于12号染色体长臂;目前认为PAH基因突变多达1100余种,大部分突变位于外显子5至外显子12之间,其中编码PAH蛋白核心功能区的外显子7突变约占25%;不同地区PAH基因突变位点各不相同,在中国以第7外显子中 c.728G>A突变率最高[3,4]。成人PKU在临床上非常少见,以往报道的成人PKU患者多表现为智力减退、鼠臭味尿液、癫痫、失语等症状,其临床表型重且预后差。本例患者与此并不相同,患者自幼智力与同龄人相比无明显差异,无鼠臭味尿液及毛发色泽改变,成年后仅表现为发作性运动伴感觉障碍,症状轻微,病史长达6个月,与大多数PKU患者表现不符。研究表明,PKU患者的临床表型及严重程度受基因型和自身代谢水平的影响,其中基因型起主导作用[5,6]。本例患者PAH基因检测示第7外显子c.722、c.728杂合变异,分别导致PAH的第241号氨基酸由精氨酸变为组氨酸、第243号精氨酸变为谷氨酰胺,为双重杂合错义突变,其致病性均已见文献报道[7,8]。这与国外报道的成人PKU突变位点不同,但符合中国的PKU常见的突变类型。临床研究表明,当存在双重突变时,疾病严重程度一般取决于相对较轻的基因突变,并且与单一纯合突变相比,严重程度相似的两种突变可能会产生更轻微的表型[9],这或许能解释该患者存在两种基因突变,但是其临床表型轻微。同时,苯丙氨酸摄取能力和自身代谢动力学的个体间差异也是导致不同临床表型的原因。血清及大脑中增高的苯丙氨酸与运动障碍的发生密切相关,苯丙氨酸会竞争性抑制酪氨酸、色氨酸等中性氨基酸在血脑屏障间的传递,影响脑内单胺神经递质如多巴、5-羟色胺等合成,递质间平衡失调造成乙酰胆碱系统相对亢进[10,11],从而导致该患者出现肢体抖动。随着病程的进展,患者出现麻木、酸痛感,结合肌电图结果分析,可能与大脑白质中感觉神经纤维髓鞘损伤有关。需要特别注意的是本例患者伴有高同型半胱氨酸血症,该患者基因检测并未发现同型半胱氨酸代谢相关酶基因异常,临床研究表明大约有30%的PKU伴有亚临床维生素B12缺乏症,而维生素B12对同型半胱氨酸代谢至关重要,因此我们猜测同型半胱氨酸增高可能是由于体内氨基酸代谢障碍及营养紊乱如缺乏维生素B12、叶酸等所致[12,13]。Wang等[14]曾报道过1例成人PKU合并高同型半胱氨酸血症,提出两种代谢紊乱是相互影响的,每一种都有可能诱发或恶化另一种代谢紊乱的表现,因此积极控制同型半胱氨酸的水平对于治疗PKU具有关键的作用。
本例患者颅脑MR表现为颅内对称性脑白质异常信号影,需要注意与中枢神经系统脱髓鞘疾病如多发性硬化、遗传性肾上腺脑白质发育不良等相关疾病鉴别[15]。该患者表现为发作性症状,但是症状持续时间及缓解时间与多发性硬化均不相符,颅内异常信号呈对称性且无脑干、小脑、脊髓等其它损害,并且结合脑脊液检查、基因检测结果分析可加以鉴别。普遍认为,PKU的脑白质损害是永久性脱髓鞘或髓鞘化延迟,但Bick等[16]认为是可逆性髓鞘结构改变,即髓鞘的改变可经过治疗后恢复的,目前这两种说法仍存在争议。因此,针对于PKU患者,定期复查影像学检查是非常有必要的,然而本例患者随访时因个人原因拒绝复查颅脑MR。
欧洲PKU诊断和治疗指南认为饮食疗法最佳状态是维持血液中苯丙氨酸浓度在120~360 μm范围之内[17],因此部分PKU患者可以不需要饮食治疗。本例患者血清苯丙氨酸浓度为216.97 μm,归类于轻度高苯丙氨酸血症[18],但是该患者的临床症状逐渐进展,且合并同型半胱氨酸血症,在治疗上,我们积极给予低苯丙氨酸饮食、叶酸、维生素B12等治疗后患者症状明显缓解。值得注意的是,PKU患者长期饮食控制有利于改善神经系统症状,但常导致患者体内维生素、矿物质等营养物质缺乏及引起心理或神经精神障碍,影响机体的生长发育,因此在低苯丙氨酸饮食过程中应注意补充所缺乏的营养物质,同时需优化心理健康[19]。
PKU是可治的遗传病之一,早期诊断、早期治疗是改善预后的关键。成人PKU患者临床症状表现不典型,尤其出现中枢神经系统损害时与脱髓鞘疾病较难区分,需结合串联质谱法、尿蝶呤谱分析以及基因检测等试验进一步明确诊断及分型[20]。该病例报道旨在提高神经科医师对PKU的认识,临床上如有不明原因的运动、感觉障碍且合并对称性脑白质损害时应该考虑到PKU的可能,应尽早完善相关检查、及时治疗,从而改善患者预后。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
昆明医科大学学报(2021年1期)2021-02-07
中国生殖健康(2020年4期)2021-01-18
中华养生保健(2020年5期)2020-11-16
中华养生保健(2020年4期)2020-11-16
医学新知(2019年4期)2020-01-02
中国社区医师(2019年12期)2019-08-26
中国医药导报(2017年15期)2017-07-05
大众健康(2017年1期)2017-04-13
