
高良姜叶绿体基因组测序与特征分析
2021-02-22 07:31黄琼林
热带作物学报 2021年1期
黄琼林
摘 要:为了探究高良姜的叶绿体基因组特征及其系统进化发育关系,本研究以高良姜总DNA为材料,采用NovaSeq高通量测序平台进行高良姜叶绿体基因组测序,并基于生物信息学方法进行高良姜叶绿体基因组的图谱构建及注释分析。结果表明:高良姜叶绿体基因组全长162 137 bp,呈典型的环状四段式结构,包括87 264 bp的大单拷贝区、15 349 bp的小单拷贝区以及2个29 762 bp的反向互补重复区;共编码132个基因,其中蛋白编码基因86个、核糖体RNA基因8个以及转运RNA基因38个。高良姜叶绿体基因组密码子偏好性较弱,偏向于以A/T碱基结尾。碱基替换分析表明,高良姜叶绿体基因组中大多数编码基因的碱基替换没有引起氨基酸的改变。基于20种物种叶绿体基因组的系统发育分析发现,高良姜与同属植物艳山姜、益智的亲缘关系更近。本研究获得了高良姜的叶绿体基因组特征信息,为高良姜资源保护、遗传进化和品种选育奠定了基础。
关键词:高良姜;叶绿体基因组;密码子偏好性;碱基替换分析;系统发育分析
中图分类号:S813.3 文献标识码:A
Abstract: To explore the chloroplast genome features and phylogenetic relationship of Alpinia officinarum Hance, total DNA of A. officinarum was used to sequence by NovaSeq, the high-throughput sequencing platform. And then the chloroplast genome of A. officinarum was annotated and analyzed based on bioinformatics. The results showed that the chloroplast genome of A. officinarum exhibited a typical four-stage structure with a length of 162 137 bp and was composed of a large single-copy region (87 264 bp), a small single-copy region (15 349 bp) and two inverted repeat regions (29 762 bp). A total of 132 genes were annotated in the chloroplast genome of A. officinarum, including 86 protein-coding genes, 8 rRNA genes and 38 tRNA genes. The codon preference in the chloroplast genome was weak, and the codon tended to end with A/T bases. The codon replacement in the majority of coding genes did not lead to the alteration of amino acids. Additionally, phylogenetic analysis in the chloroplast genome of 20 species revealed A. officinarum shared closer relationship with A. zerumbet and A. oxyphylla. Collectively, this study reported the characteristic information of A. officinarum chloroplast genome, which would provide a firm foundation for the researches on genetic evolution and variety breeding of A. officinarum.
Keywords: Alpinia officinarum; chloroplast genome; codon preference; base replacement; phylogenetic analysis
DOI: 10.3969/j.issn.1000-2561.2021.01.001
高良姜(Alpinia officinarum Hance)为姜科山姜属多年生草本植物,主产于广东、广西和海南,其根茎入药可散寒止痛,温中止呕,是我国传统大宗中药材之一。目前,高良姜野生资源已基本灭绝,栽培高良姜由于种植强度大、成本高、收益低以及田间管理困难,药农种植积极性降低,导致高良姜栽培面积和产量显著下降,一些地区已经出现高良姜供不应求的局面[1-2]。因此,加强高良姜种质资源保护和可持续利用研究是当前高良姜产业的重要课题之一。目前主要集中于高良姜的化学成分和药理活性等方面的研究,而遗传多样性、进化和发育等方面的研究则较少。
叶绿体是植物细胞进行光合作用的场所,能为植物提供生命活动所必需的能量[3]。葉绿体拥有相对独立的遗传体系,包含一个呈环状、结构稳定的基因组,即叶绿体基因组[3-4]。与庞大的核基因组相对,叶绿体基因组DNA分子较小,长度一般在115~165 kb之间[5]。由于保守程度高、进化速率适中,叶绿体基因组已被广泛应用于植物鉴定、系统发育及遗传进化分析等研究[6-8],基于叶绿体的基因工程在种质资源保护、品种优育中发挥着越来越重要的作用。目前,益智[9]、苦参[10]、大黄[11]等常用中药材的叶绿体基因组已被相继报道。
本研究拟通过高良姜叶绿体基因组测序,从密码子偏好性、进化发育关系等方面分析高良姜叶绿体遗传体系,以期为高良姜的遗传背景、进化发育提供叶绿体基因组信息,促进高良姜种质资源保护和基因工程研究奠定基础。
1 材料与方法
1.1 材料
高良姜新鲜植物采自广东省湛江市徐闻县,液氮速冻后于?80 ℃保存备用。经岭南中药资源教育部重点实验室(广州中医药大学)詹若挺研究员鉴定为高良姜(Alpinia officinarum Hance)。
1.2 方法
1.2.1 DNA提取、文库构建及测序 取100 mg高良姜叶片,采用改良CTAB法提取总DNA,并分别使用Qubit荧光光度计、Nanodrop分光光度计和1%琼脂糖凝胶电泳检测DNA样品的浓度、纯度和完整性。DNA样品检测合格后,经超声破碎随机打断,采用NexteraXT DNA Library Preparation Kit构建DNA文库,质检合格后采用NovaSeq测序平台进行高良姜叶绿体基因组测序。
1.2.2 序列拼接、注释和图谱绘制 将测序所得的原始读序(raw read)用NGS QC Tool Kit软件包进行数据质量检测和过滤,去除低质量序列、接头序列及含有不确定碱基序列,以得到高质量序列(clean read),随后使用SPAdes 3.11.0软件对所得的clean read进行序列拼接,参考物种及序列为益智(Alpinia oxyphylla, NC_035895),并验证拼接序列是否连接成环,序列以Fastq格式储存。采用PLANN软件对高良姜叶绿体基因组全序列进行编码基因注释。根据注释结果,使用OrganellarGenomeDraw(OGDRAW)软件绘制高良姜叶绿体基因组图谱。将序列数据和基因注释信息上传到GenBank数据库,获得序列登记号为MT254526。
1.2.3 密码子偏好性分析 采用CodonW v1.3软件对高良姜叶绿体转录组的密码子组成进行分析,获得有效密码子(effective number of codon, Nc)和相对同义密码子使用情况(relative synonymous codon usage, RSCU)等参数,评价高良姜叶绿体转录组的密码子使用偏好性。其中,Nc取值范围为20~60,该值越大,密码子使用的随机性越大,使用偏性越小。RSCU值则以1为限,若RSCU>1,该密码子使用频率较高;若RSCU<1,该密码子使用频率较低。
1.2.4 同义替换和非同义替换分析 将高良姜与参考物种益智等的叶绿体基因组序列导入MAFFT v7.429软件进行比对,寻找差异碱基,并使用PAML v4.9软件计算同义替换率Ks、非同义替换率Ka值,以评价高良姜叶绿体基因组基因的进化情况。其中,Ks=同义替换SNP数/同义替换位点数,Ka=非同义替换SNP数/非同义替换位点数。同义替换是指碱基的变化没有导致氨基酸的突变,而非同义替换指碱基的变化导致了氨基酸的突变。
1.2.5 系统发育关系分析 从GenBank数据中下载益智(Alpinia oxyphylla, NC_035895)、艳山姜(Alpinia zerumbet, JX088668)、白豆蔻(Amomum krervanh, MF991963)、黄花姜黄(Curcuma flaviflora, NC_028729)、观音姜(Curcuma roscoeana, NC_022928)、蜂巢姜(Zingiber spectabile, NC_020363)、美葉闭鞘姜(Costus pulverulentus, KF601573)、单花姜(Monocostus uniflorus, KF601572)、翅果竹芋(Thaumatococcus daniellii, KF601575)、豹纹竹芋(Marania leuconeura, KF601571)、美人蕉(Canna indica, KF601570)、旅美人蕉(Ravenala madagascariensis, NC_ 022927)、流苏兰花蕉(Orchidantha fimbriata, KF6015969)、粉鸟蝎尾蕉(Heliconia collinsiana, NC_020362)、小果野蕉(Musa acuminate, HF677508)、野蕉(Musa balbisiana, NC_028439)、蕉麻(Musa textilis, NC_022926)等单子叶植物和牛蒡(Arctium lappa, MH161419)、紫菀(Aster tataricus, MH669275)等双子叶植物的叶绿体基因组序列,与高良姜的叶绿体基因组序列一起导入MAFFT v7.429软件进行多重序列比对,然后使用IQTREE 1.6.12软件以最大似然法(maximum likelihood, ML)和1000次重复次数构建系统发育关系树。
2 结果与分析
2.1 高良姜叶绿体基因组测序质量
高良姜叶绿体基因组经测序后获得15 840 078条raw read,经过滤后得到15 763 357条clean read,有效率为99.5%。平均测序深度为203.3X,超过了序列拼接要求的100X平均测序深度。碱基正确识别率>99%的碱基占比为96.6%,碱基正确识别率>99.9%的碱基占比为91.1%,平均碱基测序错误率为0.03%。上述结果说明高良姜叶绿体基因组测序质量良好。
2.2 高良姜叶绿体基因组的结构特征
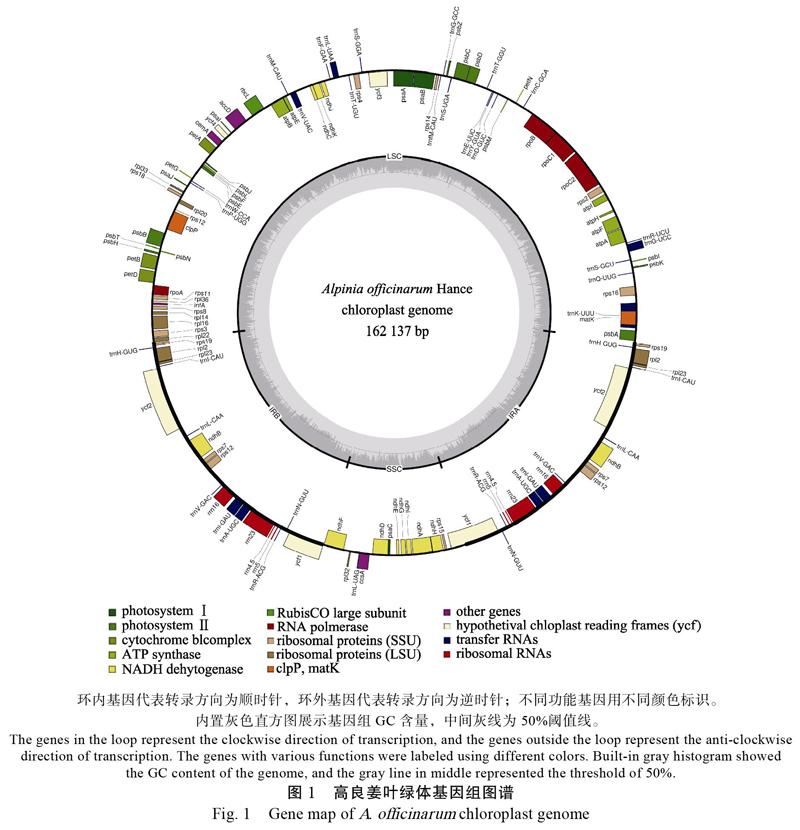
由图1可知,高良姜叶绿体基因组全长为162 137 bp,平均GC含量为36.2%,呈典型的环状四段式结构,包含始末位置为1~87 264 bp的大单拷贝区(large single-copy region, LSC),始末位置为117 027~132 375 bp的小单拷贝区(small single-copy region, SSC),以及位于LSC和SSC之间、两段反向互补重复的反向重复区(inverted repeat, IR)IRA和IRB,始末位置分别为87 265~ 117 026 bp和132 376~162 137 bp。
高良姜叶绿体基因组共编码132个基因,归属为三大类,包括86个蛋白编码基因(protein coding gene, PCG)、8个核糖体RNA(ribosomal RNA, rRNA)基因和38个转运RNA(transfer RNA, tRNA)基因。其中trnK-UUU、rps16、trnG-UCC、atpF、rpoC1、trnL-UAA、trnV-UAC、petB、petD、rpl16、rpl2、ndhB、trnI-GAU、trnA-UGC、ndhA基因各含有1个内含子,clpP、ycf3基因含有2个内含子,rps12基因存在反式剪接情况。
2.3 密码子偏好性分析
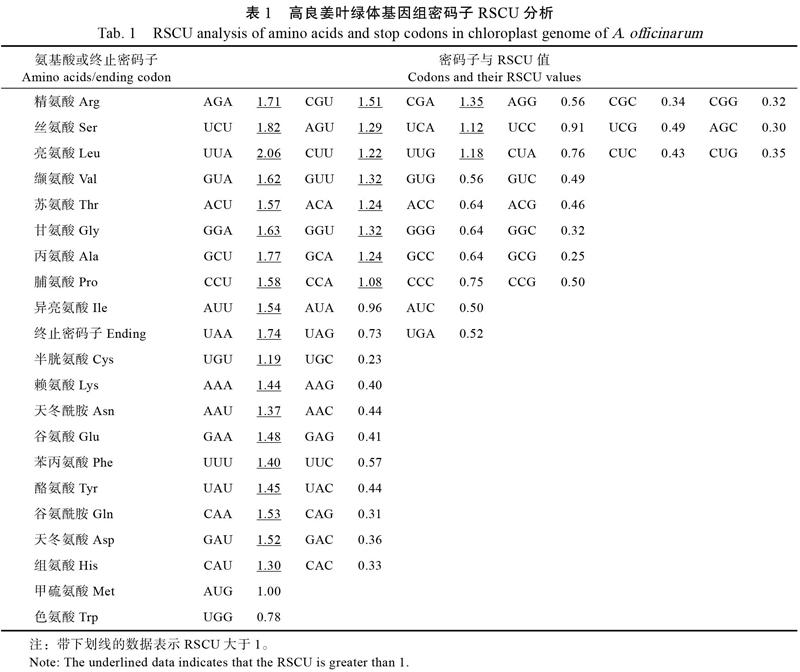
密码子组成分析结果显示,高良姜叶绿体基因组的Nc值为46.18,说明其密码子偏好性较弱。高良姜叶绿体基因组密码子的第3位上的GC碱基含量GC3s为24.27%,GC含量为37.53%,均小于50%,说明高良姜叶绿体基因组的密码子使用更倾向于A和T碱基。高良姜叶绿体基因组RSCU分析如表1所示,在64种密码子中,RSCU值大于1的密码子有30个,其中以A或T碱基结尾的密码子有29个,表明高良姜叶绿体基因组的密码子偏向以A或T碱基结尾。
2.4 同义替换和非同义替换分析
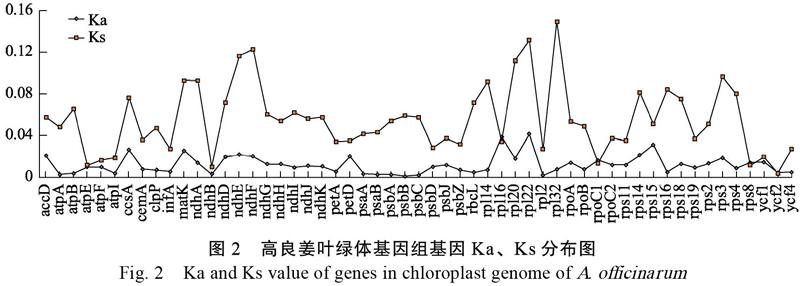
由图2可知,高良姜叶绿体基因组中共有55个基因存在碱基变化,其中大部分基因的Ks值> Ka值,即Ka/Ks<1,说明它们的进化受纯化选择。而rpl16、rpoC1、rps8、ycf2基因的Ka/Ks>1,说明这些基因的进化有正选择效应。
2.5 系统发育分析
将高良姜与17种单子叶物种的叶绿体基因组进行比对,并以牛蒡和紫菀2种双子叶植物为参照,采用最大似然法(maximum likelihood, ML)进行系统进化分析,以便理解高良姜的进化位置。结果如图3所示,高良姜与同为姜科山姜属的艳山姜、益智最先聚集在一起,说明它们的亲缘关系最近,与蕉麻等芭蕉科物种的亲缘关系则较远。
3 讨论
高良姜是药食两用的药材,市场需求量大,其植物资源及产品开发可产生较好的社会和经济效益。开展高良姜的叶绿体基因组测序及分析,可有效探讨高良姜在姜科以至单子叶植物中的系统发育关系和演化位置,为高良姜光合作用调控、抗性条件筛选等研究奠定基础,进而推动基于叶绿体基因组的高良姜分子育种和品种优育研究,以期达到高良姜资源可持续利用的目的。
本研究以基因组DNA为材料,采用高通量测序技术进行高良姜叶绿体全基因组测序,并通过生物信息学对测序数据进行编辑和分析,获得了高良姜叶绿体基因组图谱及其编码基因注释。与传统方法相比,本研究所用的方法既避免了叶绿体DNA分离等高难度实验,也不需要设计特异性引物对叶绿体基因组进行PCR扩增,具有实验周期短、劳动强度小、结果信息丰富等优势。
本研究测得高良姜叶绿体基因组全长为162 137 bp,具有高等植物叶绿体基因组的典型环状四段式结构,包括87 264 bp的大單拷贝区、15 349 bp的小单拷贝区以及2个29 762 bp的反向互补重复区,这些区域的长度与被子植物叶绿体基因各组分长度范围基本一致[12]。高良姜叶绿体基因组编码132个基因,包括蛋白编码基因86个、rRNA基因8个及tRNA基因38个,其基因组成与益智[9]等山姜属植物的叶绿体基因组类似。系统发育分析结果表明,高良姜首先与同是山姜属的艳山姜、益智聚在一起,然后再与豆蔻属白豆蔻、姜属蜂巢姜、姜黄属黄花姜黄和观音姜聚集,形成了姜科分支;而其他来源于闭鞘姜科、竹芋科、美人蕉科、旅人蕉科和芭蕉科等单子叶植物聚成另一分支。上述单子叶植物与参照的双子叶植物的牛蒡、紫菀也明显区分。在整个系统发育树中,相同科属的物种聚在一起,聚类效果较好,能较好地反映高良姜在姜科及单子叶植物中的进化关系。
在许多物种中存在着某一或几种特定的密码子使用频率高于其同义密码子的现象,这种现象被称为密码子偏好性。密码子的偏好性可反映基因以至物种的起源和进化方式,对基因功能及其编码蛋白的表达也有影响[13]。本研究结果发现,高良姜叶绿体基因组的Nc值为46.18,该值在20~60取值范围中更偏向60,说明高良姜叶绿体的密码子偏好性较弱,密码子的使用频率没有太大的差异。高良姜叶绿体基因组的GC含量和GC3s含量均小于50%,说明基因组在碱基组成上A和T的占比更高,且密码子也主要以A或T结尾。
由于密码子的简并性,有64种碱基组合编码20种氨基酸,因此DNA碱基的替换对编码氨基酸造成2种结果,即同义替换和非同义替换。前者不改变编码氨基酸,后者则改变了编码氨基酸。非同义替换(Ka)与同义替换(Ks)的比率是揭示进化率和自然选择压力的重要指标[14]。一般而言,大多数蛋白的同义替换发生率高于非同义替换发生率[15],即Ka/Ks<1。本研究也发现高良姜叶绿体基因组51个编码基因的Ka/Ks<1,占到发生碱基替换基因总数的92.7%。本研究还发现,rpl16、rpoC1、rps8、ycf2基因的Ka/Ks比值大于1,说明这些基因的进化表现出正选择效应,提示其可能处于快速进化的状态。研究这些正选择效应基因对高良姜以至山姜属物种的进化有着积极的意义。
本研究通过高通量测序技术完成了高良姜叶绿体基因组测序和图谱构建,并分析其密码子偏好性、碱基替换以及进化发育关系,为后续基于叶绿体基因组的高良姜遗传进化、变异及育种研究提供了参考。
参考文献
谭业华, 陈 珍. 南药高良姜生产调查、问题分析及发展对策[J]. 海南师范大学学报(自然科学版), 2014, 27(3): 293-296.
杨 全, 严寒静, 庞玉新, 等. 南药高良姜药用植物资源调查研究[J]. 广东药学院学报, 2012, 28(4): 382-386.
Wang X, Zhou T, Bai G, et al. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships[J]. Scientific Reports, 2018, 8(1): 12379.
Tian N, Han L, Chen C, et al. The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species[J]. PLoS One, 2018, 13(3): e192956.
Zhang Y, Du L, Liu A, et al. The complete chloroplast genome sequences of five epimedium species: Lights into phylogenetic and taxonomic analyses[J]. Frontiers in Plant Science, 2016, 7: 306.
Zhou Y, Nie J, Xiao L, et al. Comparative chloroplast genome analysis of rhubarb botanical origins and the development of specific identification markers[J]. Molecules, 2018, 23(11): 2811.
Shen X, Guo S, Yin Y, et al. Complete chloroplast genome sequence and phylogenetic analysis of aster tataricus[J]. Molecules, 2018, 23(10): 2426.
Choi K S, Kwak M, Lee B, et al. Complete chloroplast genome of Tetragonia tetragonioides: Molecular phylogenetic relationships and evolution in Caryophyllales[J]. PLoS One, 2018, 13(6): e199626.
Gao B, Yuan L, Tang T, et al. The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family[J]. PLoS One, 2019, 14(6): e218817.
喬永刚, 贺嘉欣, 王勇飞, 等. 药用植物苦参的叶绿体基因组及其特征分析[J]. 药学学报, 2019, 54(11): 2106-2112.
左文明, 曾 阳, 杨春芳, 等. 基于高通量技术的唐古特大黄叶绿体全基因组测序及应用研究[J]. 中草药, 2019, 50(22): 5545-5553.
张同武. 植物细胞器基因组测序,组装及比较基因组学研究[D]. 杭州: 浙江大学, 2012.
叶友菊, 倪州献, 白天道, 等. 马尾松叶绿体基因组密码子偏好性分析[J]. 基因组学与应用生物学, 2018, 37(10): 4464-4471.
Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models[J]. Molecular Biology and Evolution, 2000, 17(1): 32-43.
Makalowski W, Boguski M S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2820 orthologous rodent and human sequences[J]. Proceedings of the National Academy of Sciences, 1998, 95(16): 9407-9412.
责任编辑:黄东杰
