
Eyes of differing colors in Alvinocaris longirostris from deep-sea chemosynthetic ecosystems: genetic and molecular evidence of its formation mechanism*
2021-02-22 02:00QianXINMinHUIChaolunLIZhongliSHA
Qian XIN , , Min HUI , , Chaolun LI , Zhongli SHA ,
1 Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China
2 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
3 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
4 University of Chinese Academy of Sciences, Beijing 100049, China
A bstract Coloration is an important phenotypic trait for multiple adaptive functions. It is interesting to find white-eye (AW) and orange-eye (AO) phenotypes in the shrimp Alvinocaris longirostris inhabiting the deep-sea cold seep and hydrothermal vent areas of the northwestern Pacific. By comparative transcriptome analyses, 1 491 differentially expressed genes (DEGs) were identified between AW and AO. Among them, many DEGs were associated with immunity, antioxidation, and detoxification. Two significant enzyme encoding genes, xanthine dehydrogenase, and tryptophan oxidase involved in pigment biosynthesis pathways were up-regulated in AW and AO, respectively, which might be related to the differences of white and orange eye phenotypes. Moreover, single nucleotide polymorphism (SNP) calling detected that genotypes of 28 SNP distributing in 14 unigenes were completely different between AW and AO. Particularly, there were three and two non-synonymous mutations in immune genes crustin Pm5 and antimicrobial peptide, respectively. Results indicate that the difference in eye color is probably resulted from immune response to variable micro-environmental stressors encountered in the dispersal process of the shrimps, such as symbiotic microbes, pathogens, and toxic substances, and might be genetically fixed at last. The suggested pathway preliminarily explained the formation mechanism of different eye phenotypes in Alvinocaridid shrimps, providing a basis for further study on adaptive evolution of eyes in deep-sea chemosynthetic faunas. K eyword:alvinocaridid shrimps; cold seep and hydrothermal vent; differentially expressed genes; eye color; single nucleotide polymorphism (SNP) mutation; transcriptome
1 INTRODUCTION
Deep-sea cold seeps and hydrothermal vents are unique chemoautotrophic ecosystems with special physical and chemical properties: high static pressure, low oxygen concentration, enrichment of heavy metals, and accumulation of chemical toxins (Lowell et al., 1995; Brown and Hodgson, 2001). Surprisingly, numerous caridean species of the family Alvinocarididae are dominated in these chemosynthetic ecosystems (Komai et al., 2016). Due to the extremely dim light of deep-sea cold seep and hydrothermal vent environments, the eyes of Alvinocaridid shrimps degenerate in varying degrees. In the genus of blind shrimp Rimicaris, conventional anterior eyes of the adults are completely lacking (Van Dover et al., 1989; Qian et al., 2009), while another genus Alvinocaris retain the external eye structure with photoreceptor partially or completely missing (Pond et al., 1997; Charmantier-Daures and Segonzac, 1998). Among them, Alvinocaris longirostris Kikuchi and Ohta, 1995 is the only species found co-distributed in both seep and vent environments of the Northwest Pacific Ocean (Komai and Segonzac, 2005). Interestingly, two phenotypes of eyes, white-eye (AW: ~80%) and orange-eye (AO: ~20%) were discovered in this shrimp species.
Eye color has been reported to be affected by multiple environmental factors. For example, eye color mutations can be induced by radiation in the chalcid Dahlbominus (Baldwin, 1962), while in the fly Exorista japonica, eye color is temperaturesensitive, which changes from deep to pale red with increasing temperature (Ichiki et al., 2007). However, the molecular and genetic mechanisms underlying eye color mutations in crustaceans and even arthropods are poorly understood. Ommochrome (derived from tryptophan) and pteridine (derived from guanine) are two major classes of pigment in the eyes of crustaceans and insects (Elofsson and Hallberg, 1973; Summers et al., 1982). The mutations of brown, scarlet, or white gene have been revealed resulting in the eye color changes of the fly Drosophila melanogaster, by means of directly impeding the assembly or transport of the xanthommatin and pteridine pigments (Ferré et al., 1986; Besansky and Fahey, 1997; Yang et al., 2006). It also has been found that a variety of gene mutations ( prune, purple, raspberry, and rosy) can influence the content and activity of key enzymes involved in the pigment synthesis pathways, which leads to changes in pigment content and in turn influences the compound eye color (Wilson and Jacobson, 1977; Mackay and O’Donnell, 1983; Reaume et al., 1991).
Coloration is an important phenotypic trait for multiple adaptive functions, such as species identification, camouflage, warning or threatening of predators, photoprotection and photoreception (Hubbard et al., 2010). Transcriptome studies based on next-generation sequencing technology permit rapid profiling of the genes globally and functionally expressed, which has been extensively applied to the coloration studies in the mammal, fish and insect (Croucher et al., 2013; Fan et al., 2013; Jiang et al., 2014; Zhu et al., 2016), as well as to the exploration of the adaptative mechanisms in the deep-sea cold seep and hydorthermal vent animals (Bettencourt et al., 2010; Wong et al., 2015; Hui et al., 2017; Zhang et al., 2017a, b). For A. longirostris, transcriptome of whole bodies has been sequenced, and the differences between its adaptation to cold seep and hydrothermal vent environments have been uncovered by comparative study (Hui et al., 2018).
In order to reveal the mechanisms of eye color variation in A. longirostris, transcriptome of eyes was sequenced in present study, and DEGs between AW and AO were identified and analyzed. SNPs in genes were also searched, and the candidate fixed SNP mutations between the two color groups were detected. Key genes and pathways likely involved in the eye color difference of the shrimps were characterized, and therefore the molecular and genetic bases contributing to the phenotypic differences were preliminarily illustrated. It is expected to provide new insights into the special life process of macrofaunas during their adaptation to deep-sea chemosynthetic environments.
2 MATERIAL AND METHOD
2.1 Sample collection
The samples were collected near a methane seep in the South China Sea (22°6.9′N, 119°17.1′E, depth 1 119.2 m) in September 2017. They were captured by the remotely operated vehicle (ROV) Quasar MkII on the R/V Kexue (Institute of Oceanology, Chinese Academy of Sciences, China) and put into the thermomaintaining sampler. After being on board, the eyes with different colors of adult shrimps (Fig.1) were taken off and immediately frozen in liquid nitrogen and stored at -80 °C for RNA extraction. From the sample collection by ROV to the fixation, it lasted about 2 h.
2.2 Library construction, sequencing and de novo assembly

Fig.1 Shrimp Alvinocaris longirostris with orange-eyes and shrimp with white-eyes
Due to limited number of Alvinocaris longirostris obtained, especially shrimps with orange eyes, three biological replicates were included for samples of white-eye (AW) and orange-eye (AO) shrimps, respectively. The RNA extraction, library construction, sequencing and transcriptome assembly were performed according to our previously described procedures (Hui et al., 2018). In general, 1.5 μg RNA of each eye sample was extracted with TRIzol kit (Invitrogen, USA) firstly. Six libraries were then constructed using NEBNext®Ultra™ RNA Library Prep Kit and sequenced on an Illumina HiSeqTM2000 platform producing paired-end reads with length 150 bp. To get clean reads, the raw reads were filtered by removing reads containing adapter, ploy-N (with the ratio of ‘N’ >10%), and low quality reads (bases with Q value <20 accounting for more than 40% of total bases of the read) through Perl program. Transcriptome de novo assembly was carried out by using Trinity V.2.2.1 (Grabherr et al., 2011) with default parameters, except min_kmer_cov set to four. Modules of Inchworm, Chrysalis, and Butterfly in Trinity were then used to assemble the clean sequences into contigs, de Bruijin graphs and full-length transcripts sequentially. The one with the longest length of redundant transcripts was defined as a unigene. Furthermore, the completeness and redundancy of the assembled transcriptome was evaluated by checking the coverage of the 978 conserved core genes of Metazoa (https://busco.ezlab.org/) using BUSCO 3 (Simão et al., 2015).
2.3 Gene functional annotation
All unigenes were submitted for Basic Local Alignment Search Tool (BLAST) and annotated against four databases, including the National Centre for Biotechnology Information NCBI NR (nonredundant protein sequences), SwissProt (http://www.ebi.ac.uk/uniprot), KEGG (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/), KOG (euKaryotic Ortholog Group; http://www.ncbi.nlm.nih.gov/COG/) with E-value <1E-5. Then analyses were performed to obtain the potential functions of the unigenes based on known orthologous gene products. GO (gene ontology) annotation was implemented by Blast2GO based on the NR annotation information, and GO function classification statistics for all unigenes was conducted using WEGO software (Ye et al., 2006).
2.4 Gene expression level and differential expression analyses
After obtaining the read counts for each gene by mapping the clean reads to the assembled transcriptome, the gene expression level was normalized to reads per kilobase per million (RPKM). The edgeR package (http://www.r-project.org/) was then used to identify DEGs between AW and AO with a low threshold of fold change (FC) ≥2 (|log2FC| ≥1) and P-value <0.01 in order to discover more candidate genes probably involved in the eye differentiation. GO functional analysis and KEGG Pathway analysis were further performed for the DEGs. GO enrichment was implemented by the GOseq R packages (Young et al., 2010), and GO terms with Q-value ≤0.05 were defined as significantly enriched. KEGG classification was conducted using KOBAS software (Kanehisa et al., 2008), and pathways with Q-value ≤0.05 were defined as significantly enriched.
2.5 Quantitative real-time PCR (qRT-PCR) validation
qRT-PCR was performed to verify the accuracy of expression level of the unigenes revealed by transcriptome. The experiments and statistical analysis were conducted following our described procedures (Hui et al., 2018). In total, eight pairs of primers (Supplementary Table S1) for unigenes were designed to amplify the cDNA sequences, respectively. Three replicates for each group were included. 2-ΔΔCtmethod was selected to calculate the relative expression level. The β- actin gene was used as reference gene with primers (5′-3′) GAATCCACGAGACCACCTAC and ATCCACCGATCCATACAGAG. The amplification eき ciency ( E) of the primers was estimated according to the slope of the standard curve and with the formula E=[10(-1/slope)-1]×100. One way analysis of variance (ANOVA) in SPSS 16.0 was undertaken to test the significance of difference, in which P <0.01 was set as the criterion.
2.6 SNP calling and genotyping
To determine whether the difference in eye colors is due to genetic mutations, SNP analysis was further performed. The assembled sequences of A. longirostris eye transcriptome were used as reference. SNPs were detected by mapping reads of AW and AO using BWA (http://bio-bwa.sourceforge.net/; Li and Durbin, 2009) with default setting. The software package SAMtools (http://samtools.sourceforge.net/; Li et al., 2009a) was used to convert sequence alignment/map (SAM) file to sorted binary alignment/map (BAM) file. According to the relationship between the SNP allele and the reference base, the genotypes of the samples were divided into three types constituted of homozygous mutation, heterozygous mutation, and non-mutant type consistent with the reference.
2.7 Key gene sequence analysis
To understand the potential function of genes with fixed SNPs, these gene sequences were further characterized. Open reading frames (ORFs) in gene sequence were searched using online tool ORFfinder (http://www.ncbi.nlm.nih.gov/orき nder/). Then BLASTx was performed to predicted protein coding domains. The online tools SMART (http://smart.embl.de/; Letunic et al., 2012) and signalIP (http://www.cbs.dtu.dk/services/SignalP/; Petersen et al., 2011) were used to predict protein functional domains and signal peptides, respectively.
3 RESULT
3.1 Transcriptome sequencing and assembly
Six cDNA libraries were constructed, and 157 051 666 and 182 527 846 raw reads were generated from three AW and three AO samples by sequencing, respectively. After filtering, 154 384 908 and 179 635 908 clean reads were obtained, resulting in 22.84 G (AW) and 26.61 G (AO) clean bases with Q20>98.76% and Q30>96.29% in all six samples. The clean reads were assembled into 64 352 unigenes, with length ranging from 201 bp to 43 925 bp (Supplementary Fig.S1) with an average length of 956 bp and N50 length of 1 868 bp. BUSCO analysis identified 936 (95.7%) complete BUSCOs, of which 20 (2.0%) were duplicated. All these indicated a high quality of sequencing and assembling.
3.2 Functional annotation of unigenes
In total, 21 922 (34.07%) unigenes were annotated in different databases. Specifically, 21 775 (33.84%) and 15 227 (23.66%) unigenes had significant hits in the NR database and SwissProt database, respectively. The best hit of the majority of the annotated unigenes (5 257, 24%) was Hyalella azteca sequence in the NR database. There were 2 630 (12.08%) unigenes assigned to 47 subcategories in GO analysis, mainly including ‘cellular process’, ‘metabolic process’, ‘single-organism process’, ‘cell’, ‘cell part’, ‘binding’, and ‘catalytic activity’ as in most of other studies (Supplementary Fig.S2). As for KOG cluster, 13 809 (21.46%) unigenes were classified into 25 functional categories, and genes involved in ‘Signal transduction mechanisms’ took a large proportion (35.03%) (Supplementary Fig.S3). By KEGG analysis, 10 146 (15.77%) unigenes were found involved in 233 different biological pathways, among which the largest number of unigenes were assigned to the ‘metabolic’ pathway (1 335, 26.61%), and there were 245 (4.88%) unigenes identified in the ‘Biosynthesis of antibiotics’ and ‘Endocytosis pathways’, respectively.
3.3 Differential expression genes between AW and AO and functional enrichment
Overall, 1 491 DEGs were detected, of which 493 and 998 were up-regulated and down-regulated in AO when compared with AW. The expression levels based on qRT-PCR and RNA-Seq data displayed high consistency (Supplementary Table S1), indicating the gene expression level revealed by RNA-Seq was reliable.
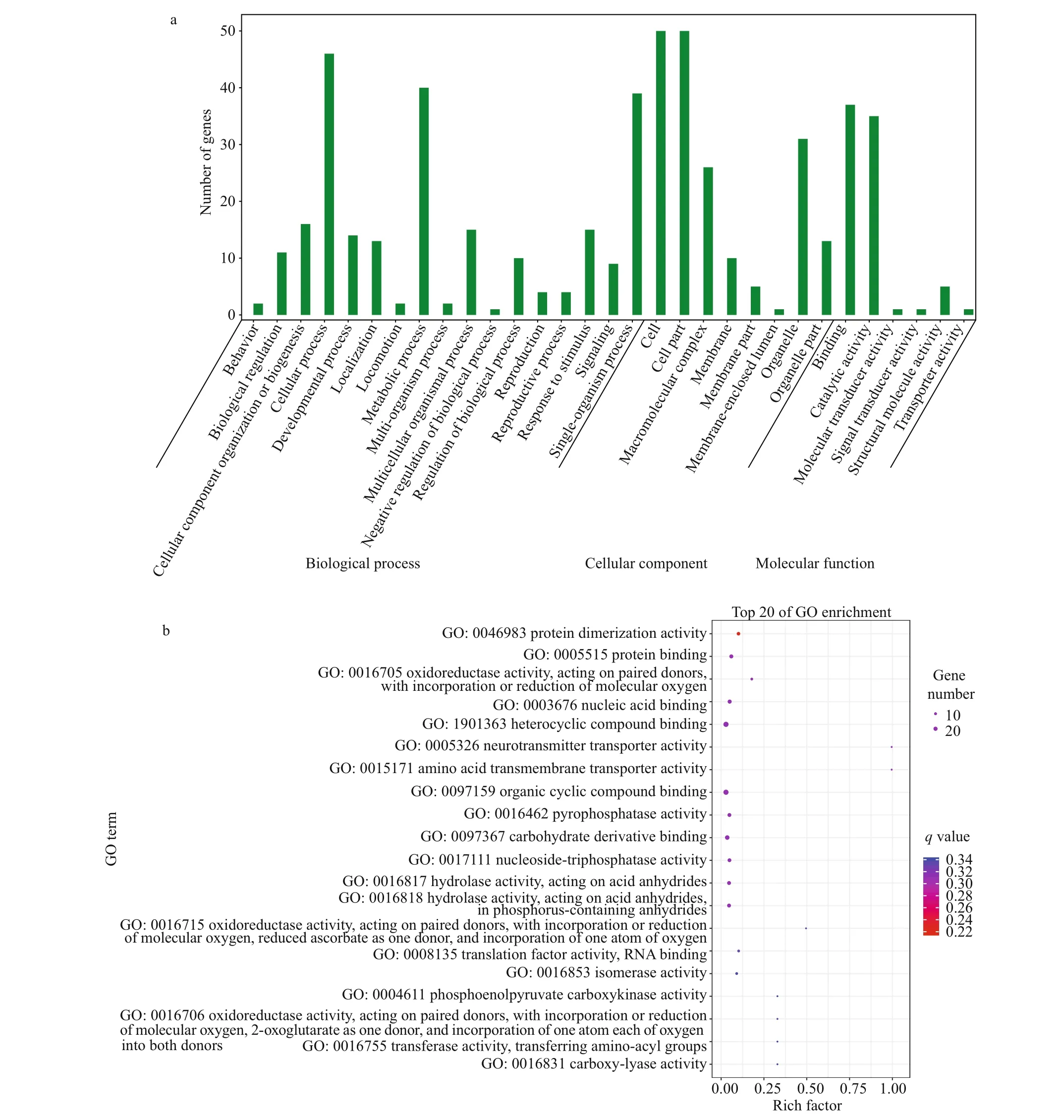
Fig.2 The overall GO enrichment categories (a) and the top 20 molecular function GO terms (b) of differentially expressed genes between white-eyes and orange-eyes of Alvinocaris longirostris
After enrichment analysis, 31 GO terms were enriched in the DEGs (Fig.2a). The GO terms of biological process described a series of events accomplished by one or more organized assemblies of molecular functions. In the top 20 enriched molecular function terms, a series of oxidoreductase activity and hydrolase activity functions were included, which were ‘oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen (GO: 0016705)’, ‘oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced ascorbate as one donor, and incorporation of one atom of oxygen (GO: 0016715)’, ‘oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, 2-oxoglutarate as one donor, and incorporation of one atom each of oxygen into both donors (GO: 0016706)’, ‘hydrolase activity, acting on acid anhydrides (GO: 0016817)’, and ‘hydrolase activity, acting on acid anhydrides, in phosphoruscontaining anhydrides (GO: 0016818)’ (Fig.2b).
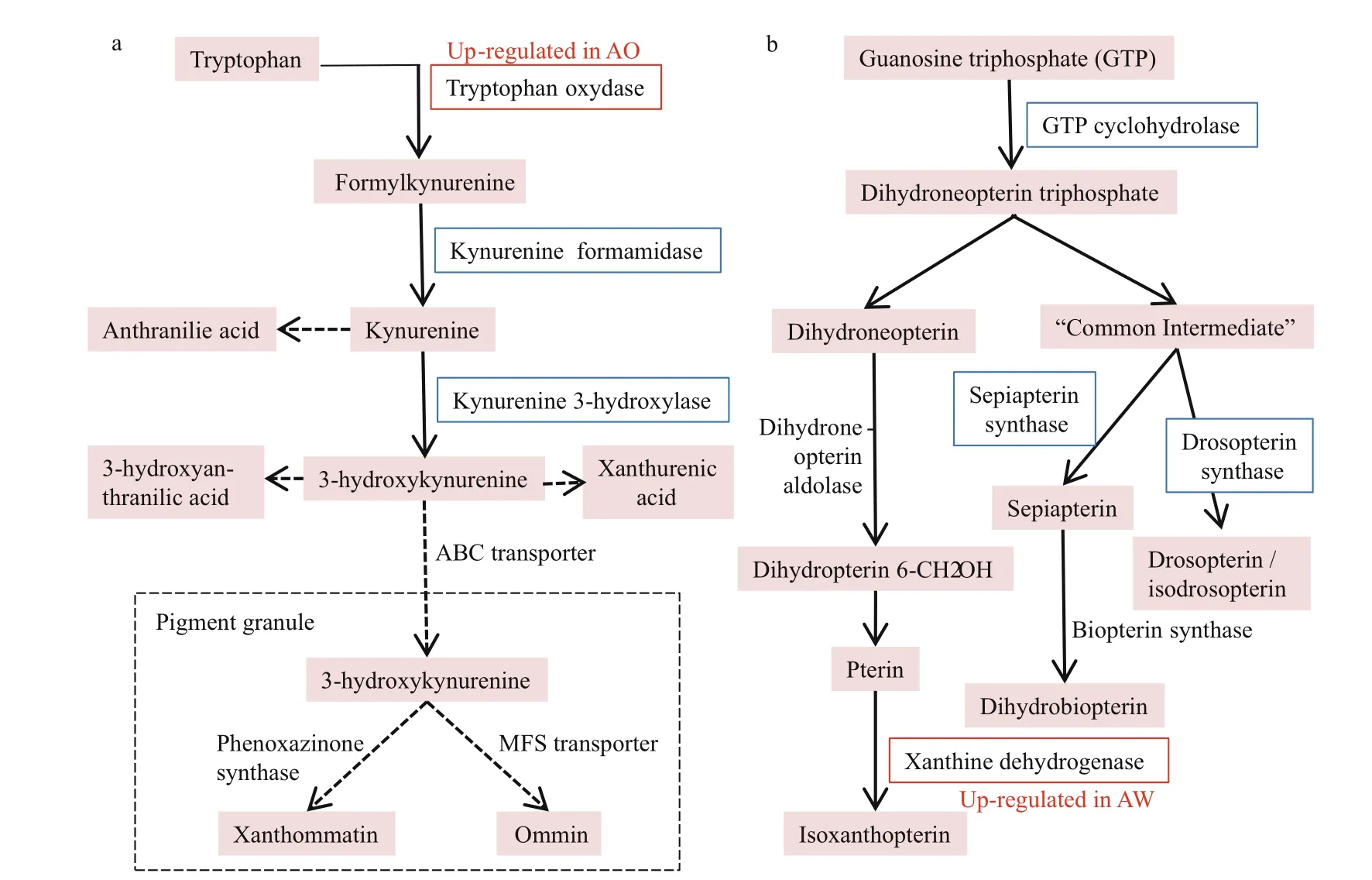
Fig.3 The two key pigment biosynthesis pathways in arthropods (revised according to Sarkar and Collins, 2000)
3.4 Stress and pigment biosynthesis related genes in the DEGs
In the DEGs between AW and AO, 22, four and one unigenes were identified to probably associate with immunity, antioxidantion and detoxification, respectively (Table 1). In the immune related DEGs, there were eight lectin s, seven crustins, four serine proteases ( SPs), one anti- lipopolysaccharide factor ( ALF), one lysozyme, and one coagulation. Unigenes related to antioxidantion included three heat shock proteins ( HSPs), and one thioredoxin ( Trx). A cytochrome b5 ( CYb5) was discovered to be differentially expressed, which might be involved in the detoxification process of the alvinocaridid shrimps. Moreover, six pigments biosynthesis related DEGs between AW and AO were identified, in which a tryptophan oxidase and three ABC transporters were up-regulated in AO, while a xanthine dehydrogenase and an ABC transporter were upregulated in AW (Table 1 & Fig.3).
3.5 SNP and fixed gene mutation identification
By direct sequencing of three AO and three AW specimens of A. longirostris, 587 689 SNPs, 21 161 insertions and 32 238 deletions were detected. After genotyping, 28 SNPs in 14 unigenes were found to be probably fixed, which were completely different between three AW samples and three AO samples (Table 2). On these SNP positions, genotypes of three AW samples were identical, while three AO samples had another same genotype. These genes containing fixed SNPs included four immune related genes ( crustin Pm5, AMP, aminopeptidase N- like, alpha- 2- macroglobulin), two structural and cytoskeletal genes ( tubulin, chitinase), and several other genes with unclear function.
4 DISCUSSION
In this study, many immunity, antioxidantion, and detoxification related genes have been revealed to be differentially expressed between AW and AO of A. longirostris, and have even yielded potentially fixed SNPs in the two eye phenotypes. Due to that A. longirostris inhabits the deep-sea chemosynthetic ecosystems, the shrimp might encounter variousstressors such as heavy metals, acidification, oxygen depletion or pathogens. Therefore, although relationship between abundances of different phenotype shrimps and habitat niches has not been surveyed, it is still supposed that the different eye colors of A. longirostris probably have been induced by environmental changes and genetic mutations might have been retained by long-term adaptive selection, presenting white- and orange-eye phenotypes.
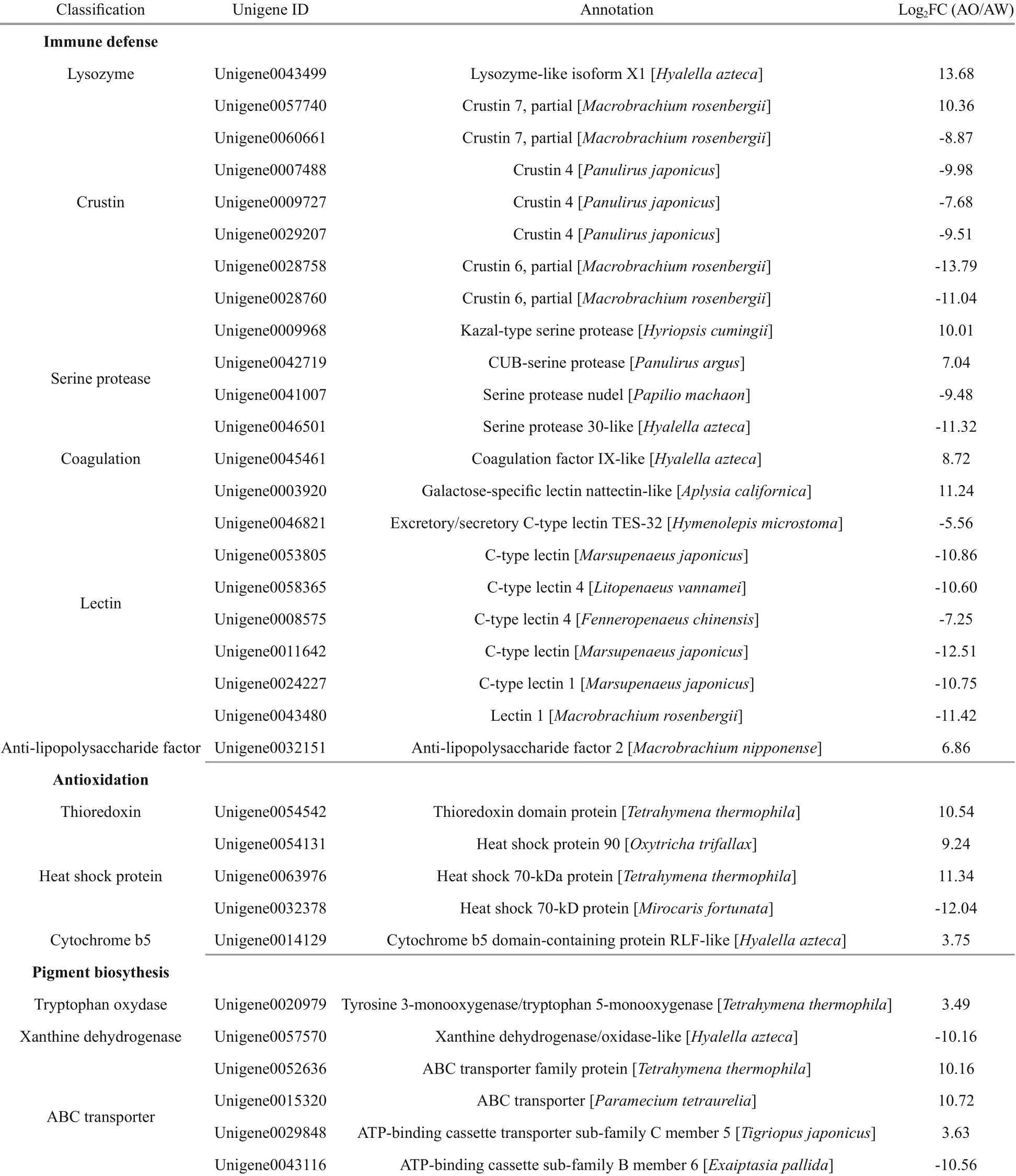
Table 1 Immunity, antioxidantion, detoxidation and pigment biosynthesis related DEGs between white-eyes (AW) and orange-eyes (AO) of Alvinocaris longirostris
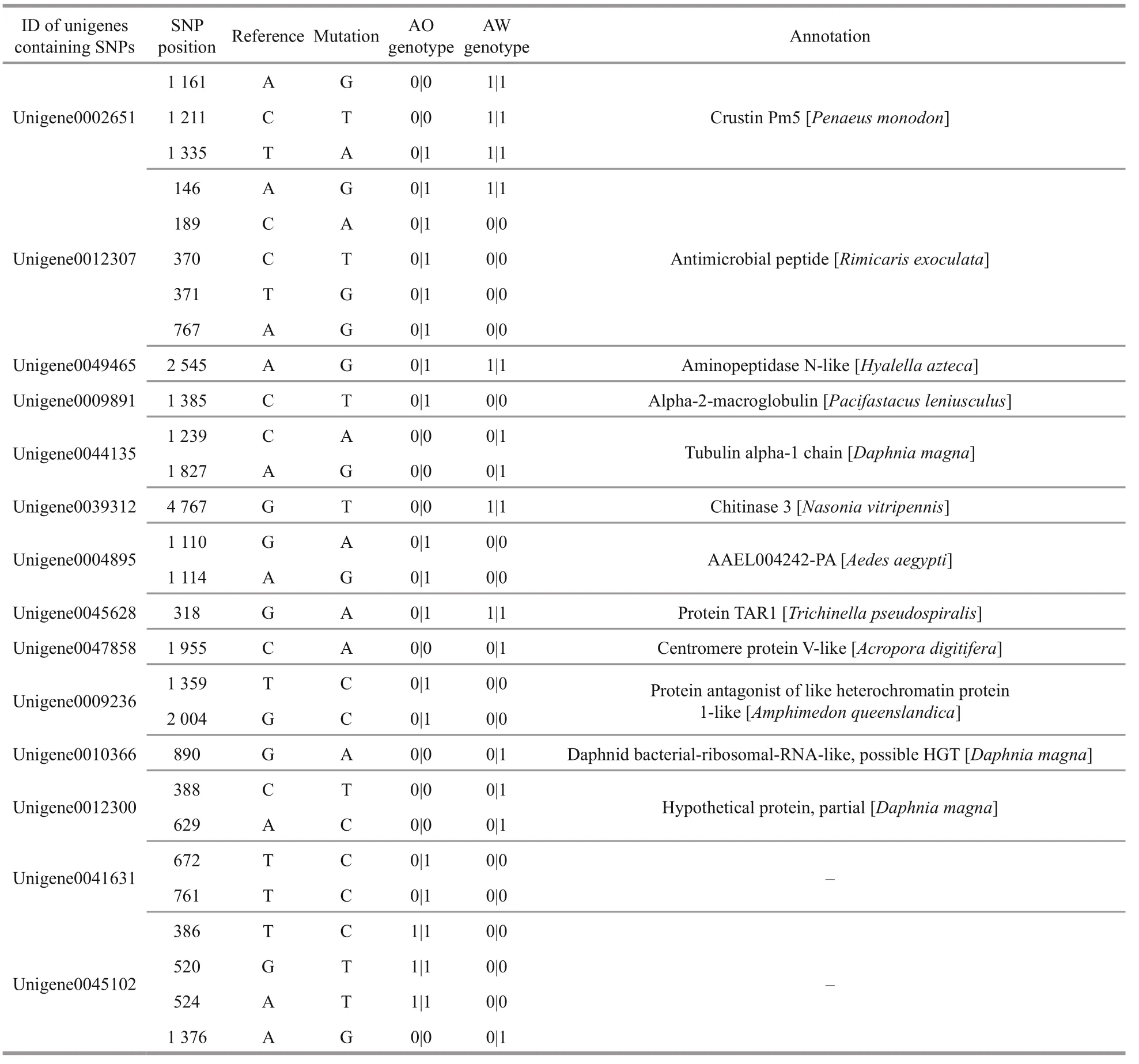
Table 2 The candidate f ixed SNPs information in Alvinocaris longirostris
4.1 Stress-related gene expression changes between different eye phenotypes
As a sensory organ, eye usually maintains the visual function, while it is also an immune privileged site that defends against invading organisms, by regulation of local immunosuppressive environment and apoptosis (Griき th et al., 1995; Hori et al., 2006; Taylor, 2007; Zamiri et al., 2007; Stein-Streilein, 2008). In this study, numerous GO terms and KEGG pathways of DEGs were discovered related to oxidation and reduction activity, signal transduction and immune response. These indicate that the AW and AO shrimps may receive different stimulations. It has been reported that eye can modulate inflammation to maximize pathogen clearance and minimize host tissue damage by innate immunity (Gregory, 2010). In total, 27 DEGs between AW and AO are identified to be associated with stress adaptation, which are generally related to immunity, antioxidantion, and detoxification. In hence, it is inferred that these eye phenotypes may be induced by the variable external environmental factors, such as various pathogens, symbiotic microbes, and toxic chemicals even in different niches of a deep-sea cold seep area. For instance, the in situ detection to the water column of South China Sea cold seeps shows that the concentration of methane, sulfide and dissolved oxygen changes with depth (Du et al., 2018), as well as changes from the center to the periphery of the cold seep vent.
4.1.1 Immunity
The innate immune system recognizes invading microorganisms through a limited number of germline-encoded pattern recognition receptors (PRRs) (Akira et al., 2006). In this study, eight lectin genes were identified in DEGs. As a kind of PRRs, lectin is able to bind to specific carbohydrate sequences, whose diversity and immune recognition functions in shrimps have been recognized (Wang and Wang, 2013). Besides, there are eight DEGs belonging to immune effector, including crustins and other ALFs that may function in protecting the organism from bacterial infections in shrimps (Pan et al., 2007; Tassanakajon et al., 2011). Moreover, a coagulation factor is discovered to be up-regulated in AO. The coagulation of hemolymph can act as the second physical barrier against the infection of pathogenic microorganisms, which effectively prevents the loss of hemolymph and promotes the healing of damage in shrimps and crabs (Perazzolo et al., 2005). Additionally, several other components involved in the innate immune response also show differentially expressed, including SPs, and lysomose. According to the previous study, different bacteria dominate different layers of water column and sediments in the same area (Wu et al., 2018), and it has been found that the components of microorganisms vary greatly as the distance from cold seep vent increases in the South China Sea (unpublished data). With high dispersal ability, alvinocaridid shrimps may encounter various microorganisms. Therefore, these results indicate that A. longirostris with white- or orange-eye may perform specific immune response when confronting diverse microbes and pathogens.
4.1.2 Antioxidantion and detoxification
Analyses also reveal that genes involved in antioxidant defense, one Trx s, and three HSPs are differentially expressed between AW and AO. Heat shock protein is a family of non-specific proteins associated with resilience and functions in renaturing heat damaged proteins, anti-oxidation and immune response (Ravaux et al., 2003, 2007; Sato et al., 2015). In deep sea cold seep and hydrothermal vent environments, sudden thermal change, hypoxia, and shortage of oxygen may lead to an increased level of reactive oxygen species (ROS) production. Even in the same cold seep area, environmental factors are also highly variable as describe above. High levels of ROS can greatly affect cellular membranes, enzymes and DNA by exerting oxidative damage to their components, such as lipids, proteins, carbohydrates and nucleotides, (Chang et al., 2009). Due to deep-sea cold seep and hydrothermal vent are naturally enriched in heavy metals (Fe, Mn, As, Cd, Cu, etc.) and dissolved gases (H2S, CH4, H2, CO2, etc.) (Gonzalez-Rey et al., 2007), the expression change of these stress resistance and detoxification related genes also indicate that the different eye phenotypes should be caused by the variable external hash environments.
4.2 Signals of immune-related gene mutations between AW and AO
Polymorphisms of the immune-related genes could affect the immune capacity of individuals to protect themselves against infection (Li et al., 2009b). In this study, SNP genotyping and analysis show that many gene mutations between the two eye groups were associated with immunity (Table 2), such as crustin Pm5 and AMP. Both of them belong to antimicrobial peptides family, a class of effective and important natural immunologically active molecules, which are present in bacteria, protozoa, invertebrates, vertebrates and plants, demonstrating a broad spectrum of activity (Bulet et al., 1999; Epand and Vogel, 1999).
In A. longirostris, the ORFs of crustin Pm5 and the AMP are 618 bp and 549 bp coding 206 and 183 amino acids, respectively, both including a signal peptide, a glycine-rich hydrophobic region, and a whey acidic protein (WAP) domain (Fig.4). Specifically, three non-synonymous mutations between AW and AO are detected in crustin Pm5 (M2L, S43N, Y60H). M2L mutation is located in the signal peptide, which could direct the nascent polypeptide from the ribosome to the endoplasmic reticulum membrane, initiating transport across the membrane, while S43N and Y60H mutations are situated within the glycine-rich region (Fig.4a). In the other AMP of A. longirostris, non-synonymous mutations between the two eye phenotypes occur, with F38S in signal peptide and E170A in the functional WAP domain (Fig.4b). The WAP domain possesses eight Cys residues that may participate in the formation of 4-disulfide core domain, which contains proteinase-inhibitory activity or antimicrobial activity or both (Couto et al., 1993). Mutations in amino acids may result in changes in antimicrobial peptides structure and function, further producingdiverse antimicrobial activities against the attack of different pathogens in crustaceans (Philip et al., 2016). It has been revealed that the antimicrobial peptide with the deletion mutants of either Gly-rich or Cys-rich regions exhibited lower antibacterial activity than the wild type crustin Pm1 in the black tiger shrimp Penaeus monodon (Suthianthong et al., 2012). Adaptation is an evolutionary process enabling an organism to fit its habitat better by means of natural selection. The genetic differentiation between immune genes of A. longirostris white- and orange-eyes may be the outcome of long-term environmental adaptive selection, especially resistance of different pathogens or symbiotic bacteria. These gene mutations should be further verified in more samples of AW and AO, and however, limited number of A. longirostris shrimps, especially shrimps with orange eyes, are available due to the diき culty of sampling in deep-sea.

Fig.4 cDNA and deduced amino acid sequences of genes crustin Pm5 (a) and antimicrobial peptide ( AMP) (b)
4.3 Pigment formation mechanism of different eye colors
The compound eyes of the arthropod, such as in insects, mainly includes two pigment biosynthesis pathways, the ommochrome biosynthesis pathway and the pteridine pigment biosynthesis pathway (Needham, 1970; Yamamoto et al., 1976; Summers et al., 1982; Sarkar and Collins, 2000). Specifically, by screening all annotated genes in the transcriptome of A. longirostris, numerous genes associated with pigment biosynthetic pathways including ommochrome and pteridines pathways are identified in the transcriptome of A. longirostris. Specifically, 11 unigenes are involved in the ommochrome anabolic pathway, including genes tryptophan 5-monooxygenase, tryptophan 2, 3- dioxygenases,
kynurenine formamidases, kynureninases, and kynurenine 3- monooxygenase (Supplementary Table S2, Fig.3a), whose expression level are all relatively higher in AO than AW. Fourteen unigenes are found related to pteridines anabolic pathway, which were GTP cyclohydrolase 1, 6- pyruvoyl tetrahydrobiopterin synthase, sepiapterin reductases, xanthine dehydrogenase/ oxidases (Supplementary Table S2, Fig.3b), and in general, the RPKMs of these genes in AO were higher than in AW, except gene sepiapterin reductase. Sepiapterin is a yellow pteridine pigment, which accumulated when lack of the enzyme sepiapterin reductase (Gao et al., 2013), which might play a significant role in the formation of AO. In addition, ABC transporters are a class of multicomponent transporters that can carry transmembrane transport of ommochrome and pteridines. Fifty ABC transporter genes are also detected from all unigenes, but most of them display no different expression between AW and AO.
In these pigment biosynthesis related genes, six are identified to be differentially expressed (Fig.3), including a tryptophan oxidase, a xanthine dehydrogenase and four ABC transporters. The tryptophan oxidase is the first key enzyme in the ommochrome biosynthesis pathway, which could change the tryptophan content and greatly influences the ommochrome synthesis pathway (Green, 1949; Baglioni, 1959; Lorenzen et al., 2002). Thus, the upregulation of tryptophan oxidase in AO may play a significant role in the synthesis of orange pigment. Meanwhile, xanthine dehydrogenase can oxidize the yellow pterdine xanthopterin to the colorless pteridine leucopterin as shown in butterflies (Watt, 1972), and mutants of the rosy ( ry) gene encoding for the enzyme xanthine dehydrogenase in Drosophila melanogaster lead to a brownish eye color phenotype reflecting a deficiency in the red eye pigment (Reaume et al., 1989, 1991). Therefore, the up-regulation of gene xanthine dehydrogenase in AW may play a critical role in the eventual appearance of white eye by converting yellow pterine to white or colorless. Additionally, ABC transporters are also closely related to the formation of compound eye pigments (Grubbs et al., 2015), by virtue of affecting the transmembrane transport of ommochrome and pterine (Ewart and Howells, 1998; Mackenzie et al., 1999). It is assumed that the two eye phenotypes might be induced by the variable environment factors in the deep-sea chemosynthetic ecosystems, such as microbes, dissolved oxygen and chemical changes, and in the long term adaptation, the stress related genes, especially immune associated genes are under selection, which may in turn affects related gene expression, the biosynthesis of eye pigments, and eventually the eye color (Fig.5). The results are expected to provide clues for the study of eye adaptation in deep-sea chemosynthetic shrimps, while the function of candidate genes and pathways will be examined in further studies.
5 CONCLUSION
In this study, comparative transcriptome for white- and orange- eyes of A. longirostris was conducted. Non-synonymous mutations were detected in two immune-related genes, crustin Pm5 and another AMP. Many genes related to immunity, antioxidation, and detoxification were identified to be differentially expressed between AW and AO. Two significant enzyme encoding genes xanthine dehydrogenase and tryptophan oxidase involved in pigment biosynthesis pathways were up-regulated in AW and AO, respectively, which may directly lead to the differences of white- and orange- eye phenotypes. In hence, the different eye color of A. longirostris are might be a response to deep-sea environment stress, such as microbes, pathogens, and toxic substances. In the long-term adaptation, the selection of stress related genes, especially immune associated genes has been fixed, which in turn affects related gene expression at molecular level, the biosynthesis of eye pigments, and finally the eye color. However, the mechanism of interrelation between immunity and pigment synthesis should be further pursued.
6 DATA AVAILABILITY STATEMENT
Raw reads of the sequencing are deposited in the Sequence Read Archive (SRA) database of NCBI with the BioProject accession number PRJNA548620. All other data generated or analyzed during this study are included in this published article and its supplementary information files.
7 ACKNOWLEDGMENT
The samples were collected by R/V Kexue. The authors wish to thank the crews for their help during collection of samples.
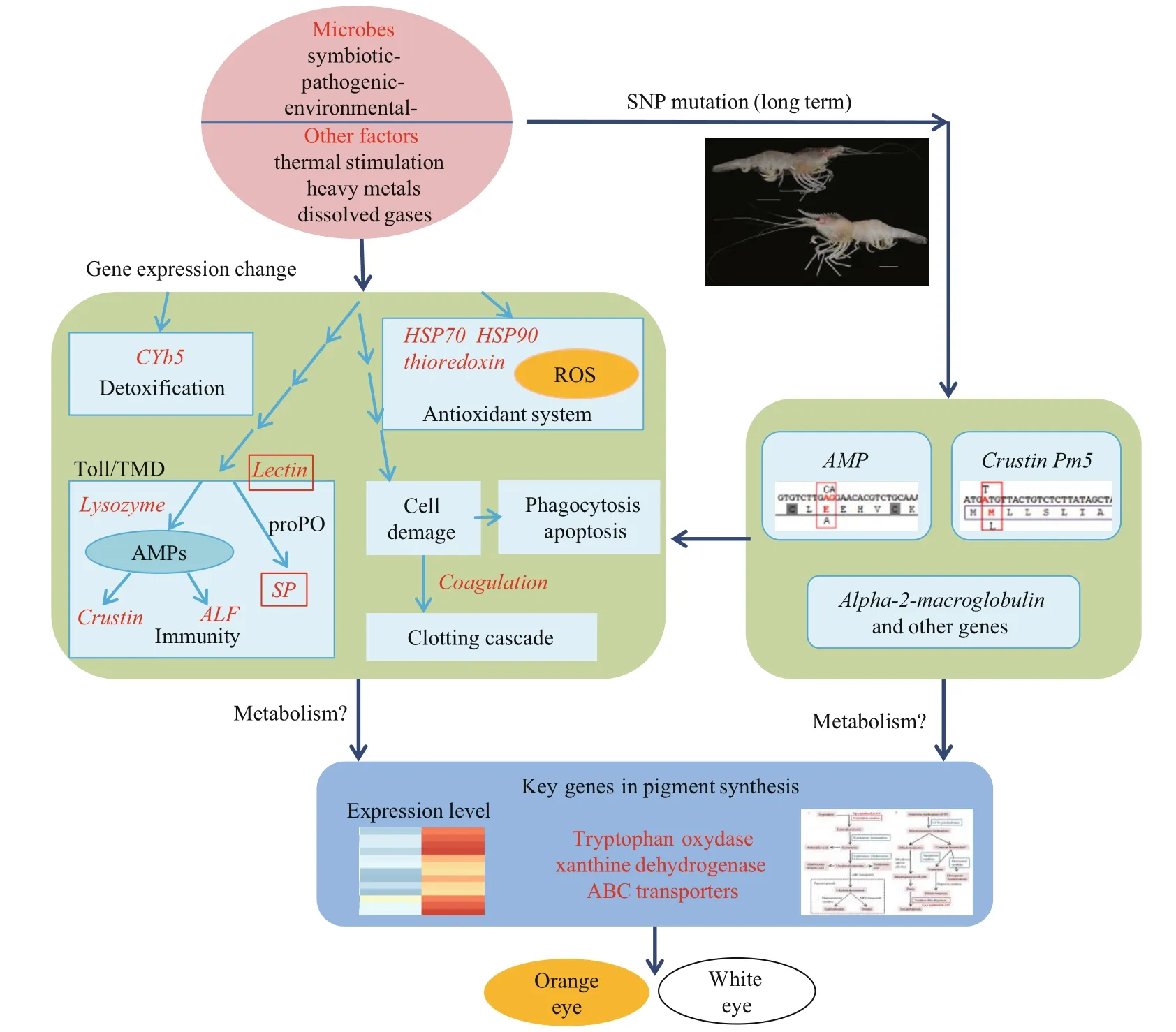
Fig.5 A putative schematic model of the molecular mechanism explaining the formation of white- and orange-eye phenotypes in Alvinocaris longirostris
 Journal of Oceanology and Limnology2021年1期
Journal of Oceanology and Limnology2021年1期
- Journal of Oceanology and Limnology的其它文章
- Influence of sequential tropical cyclones on phytoplankton blooms in the northwestern South China Sea*
- Simulated perturbation in the sea-to-air flux of dimethylsulfide and the impact on polar climate
- Performance of ecological restoration in an impaired coral reef in the Wuzhizhou Island, Sanya, China*
- Investigating factors driving phytoplankton growth and grazing loss rates in waters around Peninsular Malaysia
- Effects of oxytetracycline dihydrate and sulfamethoxazole on Microcystis aeruginosa and Chlamydomonas microsphaera*
- Reproductive cycle of Ophiopholis mirabilis (Echinodermata: Ophiuroidea) in Zhangzi Island area, northern Yellow Sea*