
Clinical characteristics and ABCC2 genotype in Dubin-Johnson syndrome:A case report and review of the literature
2021-02-22 07:21
World Journal of Clinical Cases 2021年4期
Huan Wu,Xue-Ke Zhao,Juan-Juan Zhu,Department of Infectious Diseases,The Affiliated Hospital of Guizhou Medical University,Guiyang 550001,Guizhou Province,China
Abstract BACKGROUND Dubin-Johnson syndrome (DJS) is a benign autosomal recessive liver disease involving mutations of the ABCC2 gene.It is characterized by chronic or intermittent conjugated hyperbilirubinemia,with chronic idiopathic jaundice as the main clinical manifestation.Genetic alterations of the ABCC2 gene are commonly used for diagnosing DJS;however,the causative ABCC2 point mutation in Chinese patients remains unknown.Research on ABCC2 mutations in Chinese DJS patients is extremely rare,and the diagnosis of DJS remains limited.The routine analysis of ABCC2 mutations is helpful for the diagnosis of DJS.Here,we report the clinical characteristics and ABCC2 genotype of an adult female DJS patient.This article is to expound the discovery of more potentially pathogenic ABCC2 variants will that contribute to DJS identification.CASE SUMMARY This study investigated a woman referred for DJS and involved clinical and genetic analyses.ABCC2 mutations were identified by next-generation sequencing(NGS).The patient showed intermittent jaundice and conjugated hyperbilirubinemia.Histopathological examinations were consistent with the typical phenotype of DJS.Genetic diagnostic analysis revealed an ABCC2 genotype exhibiting a pathogenic variant,namely c.2443C>T (p.Arg815*),which has not been reported previously in the domestic or foreign literature.CONCLUSION Pathogenic ABCC2 mutations play an important role in the diagnosis of DJS,especially in patients with atypical presentations.Currently,NGS is used in the routine analysis of DJS cases and such tests of further cases will better illuminate the relationship between various genotypes and phenotypes of DJS.
Key Words:Dubin-Johnson syndrome;ABCC2 genotype;Next-generation sequencing;ABCC2 mutation;Homozygous mutation;Case report
INTRODUCTION
Dubin-Johnson syndrome (DJS) is a benign autosomal recessive liver disease that is characterized by chronic or intermittent conjugated hyperbilirubinemia[1].During laparoscopy,DJS cases display a pathognomonic "black liver",which results from excessive deposition of a melanin-like pigment in the lysosomes of hepatocytes[2,3].Surprisingly,few diagnostic tools are available to detect this syndrome during liver function and physical examinations[4-7].Additionally,some DJS cases share symptoms,such as dull pain in the liver area,generalized weakness,fatigue,and loss of appetite,with other diseases,which inevitably leads to misdiagnosis[8].The main means of effective diagnosis of DJS are laparoscopic exploration and liver biopsy[9,10].However,experience in genetic diagnosis,precision treatment,and follow-up observation of DJS patients remains insufficient.
Mutations inABCC2have been observed in DJS patients since 1997[11-13],and more than 50 different mutations causing amino acid changes in the ABCC2/MRP2 protein have been described in DJS patients[12,14-16].These mutations result in a dysfunctional MRP2 protein or accelerate mRNA degradation,thus causing diminished glycosylation and impaired sorting[2,11,17].Moreover,acquired or hereditary dysfunction ofABCC2contributes to liver dysfunction,which alters bilirubin metabolism and results in hyperbilirubinemia[9,18,19].Genetic alterations of theABCC2gene are commonly used for diagnosing DJS;however,the causativeABCC2point mutation in Chinese patients remains unknown.Here,we present the case of a female DJS patient.Laparoscopic exploration and liver biopsy were used for a definitive diagnosis.Subsequently,we performed next-generation sequencing (NGS) with the aimed of identifying newABCC2mutations.
CASE PRESENTATION
Chief complaints
A 24-year-old female was reported as having "gallbladder stones and acute cholecystitis" by the local hospital,and laparoscopic cholecystectomy was performed.During the operation,the dark brown liver surface,slightly shorter edges,and a stonefilled gallbladder led to the termination of cholecystectomy (Figure 1).She was then transferred to our department for further diagnosis.
History of present illness
She is currently not ill.
History of past illness
She had not consumed alcohol or drugs hazardous to the liver,and reported no family history of liver disease.She had given birth three times and had episodes of jaundice during each pregnancy.

Figure 1 Laparoscopic images presenting a deep black-brown liver with a smooth surface and the slightly shorter edges,and a stonefilled gallbladder (orange arrows).
Personal and family history
She has no family history of liver disease.
Physical examination
The patient was conscious and stable when she arrived at our department.She presented with visible yellow stains on her skin,mucous membranes,and sclera.Her superficial features were undiluted capillaries on the face,no spider nevus in the anterior of the chest wall and neck region,and negative liver palmar.The abdomen was soft without varicose veins or exposure.The liver and spleen were non-palpable under the ribs and percussion of the hepatic region was barely painful.
Laboratory examinations
The relevant auxiliary examination involved liver function tests,routine urinalysis,immunohistochemistry tests of the blood,and a single computed tomography (CT)scan.The results of these examinations which are summarized in Table 1.
Imaging examinations
Pathological examination of the liver showed a massive deposition of pigment particles in hepatocytes,especially in the centrilobular region.These particles showed no refractive properties;therefore,they were assumed to be a biliary pigment(Figure 2A).Under the microscope,brownish-yellow pigmental granules were observed in the hepatocytes,especially in those surrounding the central vein.Furthermore,some hepatocytes showed watery and balloon-like degeneration,and nucleated inflammatory cells were present in the hepatic sinus.No obvious abnormalities were found in the portal area,fibrous tissue,bile ducts,or blood vessels between the lobules (Figure 2B).
Further diagnostic work-up
A The whole-abdomen CT scan found multiple gallbladder stones,increased liver parenchymal density,and an enlarged spleen.After excluding liver contraindications,an ultrasound-guided liver biopsy was performed for jaundice.A melanin-like pigment deposition was observed in hepatocyte lysosomes sing laparoscopy.Additionally,periodic acid-Schiff staining was negative,and reticular fiber and collagen fiber staining (Masson) showed no obvious abnormalities.We performed NGS to identify mutations in theABCC2gene.
Molecular identification
We confirmed the presence of a homozygous mutation,c.2443C>T (p.Arg815*),inABCC2in this patient (Figure 3).The mutation was located in exon 19 ofABCC2and resulted in a change from cytosine to thymine.The amino acid at position 815 of the encoded protein changed from arginine to a stop codon,leading to the premature termination of the peptide chain synthesis.This mutation is novel,and does notappear in HGMD,Esp6500siv2_ALL,or 1000g2015aug_ALL databases.In the dbSNP147 database,it is recorded as rs773850184.According to a comprehensive assessment of the clinical situation,this patient carried a pathogenic mutation inABCC2,which was the main cause of DJS.
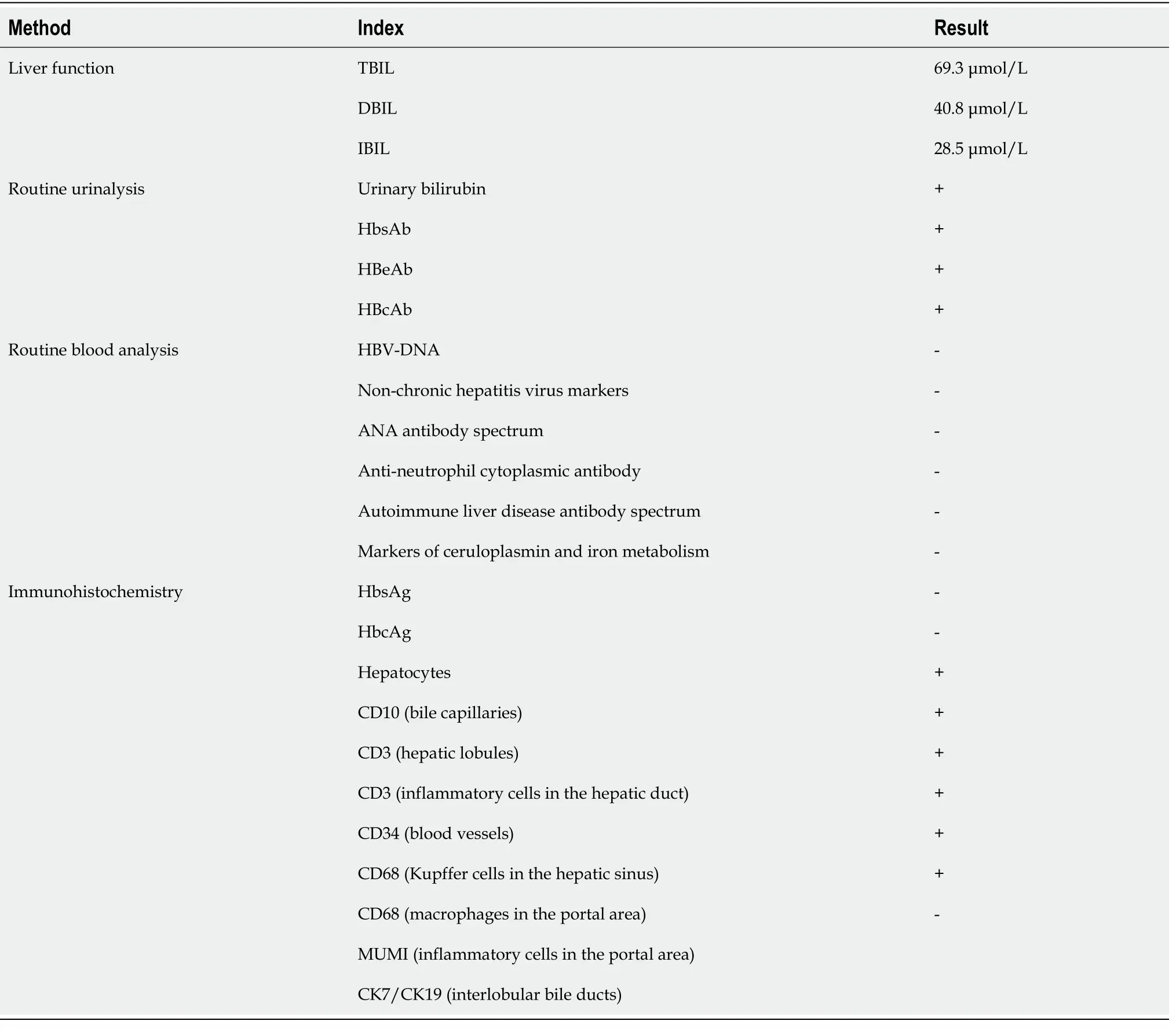
Table 1 The clinicopathological parameters of a Dubin-Johnson syndrome patient
FINAL DIAGNOSIS
The final diagnosis of the presented case is DJS.
TREATMENT
After admission,the patient was administered a light digestible diet and class I nursing.Treatment medicines included Shuganning and glutathione to protect the liver and eliminating jaundice,omeprazole to stomach acid,and hepatocyte growthpromoting hormone to promote liver cell regeneration)[20-22].Additionally,compound coenzymes,lipid soluble vitamins (II),and water-soluble vitamins were used for nutritional support.

Figure 2 Imaging results.
OUTCOME AND FOLLOW-UP
The treatment went well.The blood biochemical indices of the patient significantly improved to the following values:Total bilirubin 34.6 μmol/L;direct bilirubin,23.1 μmol/L;indirect bilirubin,11.5 μmol/L;alanine aminotransferase,11.7 U/L;aspartate aminotransferase,12.3 U/L;gamma-glutamyl transferase,11.0 U/L;alkaline phosphatase,78 U/L;cholinesterase,6454 U/L;PA,220.4 mg/L;and total bile acid,14.3 μmol/L.Blood coagulation function and routine blood analysis showed no abnormalities.The patient’s samples were submitted to Kingmed Diagnostics for mutation detection and the patient was discharged from the hospital.Further followup observation is needed for long-term prognosis.
DISCUSSION
This case of DJS had the typical features of familial conjugated hyperbilirubinemia,including elevated levels of conjugated bilirubin that developed into chronic intermittent jaundice.Pathologically,the matrix deposited with ferritin-like particles led to a black liver,with a lipofuscin like pigment deposited in the middle of the hepatic lobule.However,these symptoms are merely used as a diagnostic indicators and they lack specificity.A review of the condition showed that the patient had been pregnant three times,and that yellow staining of the systemic skin,mucosa,and sclera was present during each pregnancy.The patient was diagnosed with acute cholecystitis,and the corresponding treatment was adopted,given the pregnancy factor[23-26].The main pathological changes observed in the liver tissue were massive pigment particle deposition in hepatocytes and significant pigment granules in the central area of the lobules.It was difficult to distinguish whether the pigment particle was iron,copper,or bile pigment using hematoxylin and eosin staining.As the serum copper-blue protein level of the patient was normal,liver injury due to copper accumulation was ruled out.The pigment granules tended to have bile pigment deposits,rather than iron,as indicated by their lack of refraction.Therefore,considering the histological features and relevant clinical examination,the patient was diagnosed with DJS.The laboratory examinations performed on the patient included a liver function test,which showed increased direct bilirubin levels;a sulfobromophthalein sodium test;a urine porphyrin isomer test;an oral cholecystic contrast agent test,which showed gallbladder non-development;radionuclide hepatobiliary imaging;and a liver biopsy,which is the gold standard for the diagnosis of DJS.However,these diagnostic measures are not widely available in health-care facilities in China.As DJS is a rare hereditary disease,the lack of experience with its diagnosis and treatment brings difficulties in clinical practice.

Figure 3 Next-generation sequencing results of ABCC2 in a Dubin-Johnson syndrome patient.
With the development of sequencing technology,genetic,and epigenetic research of complex disorders is becoming more common.Therefore,multiple mutation sites have been detected in DJS patients.TheABCC2mutations identified in DJS patients include gene deletions,missense mutations,nonsense mutations,and splice site mutation.A previous study analyzing the clinical diagnosis of seven DJS patients in China found that all patients had at least one non-synonymous mutation in theABCC2gene.All identified mutations were heterozygous mutations,while only two cases had complex heterozygous mutations[2],suggesting that other clinically relevantABCC2variants were involved.Further,two new homozygous mutations,c.1013_1014delTG and 974C->G,in the eighth exon of theABCC2gene,have been identified in other countries[27-30].However,genetic analysis of DJS cases is relatively inadequate in China.In this study,the patient’s mutation was confirmed to be c.2443C>T (p.Arg815*) by sequencing,and was identified as a nonsense mutation.Therefore,a cytosine in the coding region ofABCC2was mutated to thymine,resulting in the amino acid at position 815 changing from arginine to a stop codon,which causes the premature termination of peptide chain synthesis,leading to a defective MRP2 protein.There was no report of this mutation in domestic and foreign literature databases,including China HowNet,Wanfang,Weipu,and PubMed,except in the dbSNP147 database.The DJS cases with theABCC2(p.Arg815*) nonsense mutation may contribute to potentially pathogenicABCC2variants of the genetic or metabolic liver diseases.
CONCLUSION
We reported the clinical manifestations and the methods of diagnosis of a Chinese DJS patient,who had a homozygous mutation,c.2443C>T (p.Arg815*),inABCC2.Patients with unexplained jaundice should be genotyped as early as possible,for their effective clinical management.
 World Journal of Clinical Cases2021年4期
World Journal of Clinical Cases2021年4期
- World Journal of Clinical Cases的其它文章
- Chiari malformations in children:An overview
- Effect of hospital discharge plan for children with type 1 diabetes on discharge readiness,discharge education quality,and blood glucose control
- Effect of biofeedback combined with high-quality nursing in treatment of functional constipation
- Radioactive 125I seed implantation for pancreatic cancer with unexpected liver metastasis:A preliminary experience with 26 patients
- Biliary stent combined with iodine-125 seed strand implantation in malignant obstructive jaundice
- Usefulness of prenatal magnetic resonance imaging in differential diagnosis of fetal congenital cystic adenomatoid malformation and bronchopulmonary sequestration