
儿童囊性纤维化1例及其基因型文献复习
2021-02-14 00:19陈琼华郑敬阳林洁如曾丽娥胡云婷林印涛
福建医科大学学报(社会科学版) 2021年6期
陈琼华, 郑敬阳, 林洁如, 曾丽娥, 胡云婷, 林印涛
囊性纤维化(cystic fibrosis, CF)是常见于白种人的可缩短寿命的常染色体隐性遗传病[1],属于单基因病,由位于第7号染色体的CF跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因变异导致CFTR蛋白功能障碍引起。既往认为,该病在中国人群较少见。近年来,随着对CF认识水平的提高以及基因诊断技术的发展,越来越多的CF患者被识别并诊断。中国CF儿童的基因型和表型与国外报道存在差异,有学者[2]总结了中国人群的CFTR基因突变谱,发现c.2909G>A出现频率最高,其次是c.1766+5G>T和c.263T>G。现报道1例c.1766+5G>T和c.263T>G突变所致的CF病例,并复习相关文献,以提高对该疾病的识别和诊断水平。
1 病例介绍
1.1 临床资料 患儿,女,6岁,因“反复咳嗽1个月,加剧伴发热1 d”于2019年10月入院。患儿自2岁起反复咳嗽,入院1 a前有“支气管扩张”病史,多次痰培养提示金黄色葡萄球菌和铜绿假单胞菌阳性。入院前1个月,患儿咳嗽、咳痰症状加重,当地医院予抗感染治疗10余天,咳嗽较前好转。1 d前出现发热,热峰39 ℃,于当地医院行胸片检查,提示支气管肺炎、肺大疱可能,为进一步诊治,收住笔者医院。出生史、喂养史、过敏史、生长发育史及家族史无特殊。入院查体:体温36.5 ℃,脉搏 99 min-1,呼吸30 min-1,血氧饱和度94%。神志清楚,呼吸稍促,约30 min-1,节律规整,吸气性三凹征阴性,双肺呼吸音粗,可闻及湿啰音,可见杵状指(趾)(图1),余无特殊。辅助检查:(1)血常规:白细胞22.3×109L-1,中性粒细胞比例78.1%,淋巴细胞比例14.2%,血红蛋白142 g/L,血小板530×109L-1,C反应蛋白14.27 mg/L;(2)肺部CT:双肺上叶支气管扩张(图2A,2B);(3)生化:心、肝、肾功能及电解质未见明显异常;(4)肺炎支原体抗体凝集法阳性(1∶640)、肺炎支原体IgM(ELISA法)阳性;(5)传染病筛查:乙型肝炎、丙型肝炎、梅毒、艾滋病等相关检查均阴性;(6)尿、粪常规未见异常;(7)体液免疫:IgG、IgA、IgM分别为8.50、0.92、1.47 g/L,IgE为57.60 IU/mL,补体C3、C4分别为1.17、0.31 g/L;(8)细胞免疫:各细胞百分率CD3 73%,CD3+CD8+36%,CD3+CD4+33%,CD3-CD16+CD56+17%,CD19+7%,CD3+CD4+/CD3+CD8+为0.92,大致正常;(9)支气管镜:支气管内膜炎症,支气管扩张(图2C,2D);(10)痰培养:铜绿假单胞菌;(11)液基薄层细胞制片术(肺泡灌洗液):中度炎症,以中性粒细胞为主;(12)纤毛活检:查见个别纤毛9+2微管结构异常,表现为少数联体纤毛;微管数目正常,内、外动力臂无缺失(图3);(13)肝、胆、胰、脾彩超:右肝后叶中高回声(3.3 cm×2.1 cm×1.3 cm);(14)肺通气功能:轻度阻塞性通气功能障碍。入院先后予“头孢哌酮舒巴坦钠”和“左氧氟沙星”抗感染,“盐酸氨溴索”祛痰,“布地奈德、沙丁胺醇、乙酰半胱氨酸”雾化解痉化痰,“阿奇霉素”抗支原体等治疗,行两次支气管肺泡灌洗,完善纤毛活检。治疗16 d,患儿咳嗽咳痰症状改善,体温正常,复查感染指标正常,但痰培养仍可见铜绿假单胞菌,予办理出院。

A:患儿杵状指;B:患儿杵状趾。图1 患儿杵状指(趾)Fig.1 Clubbed-finger of the Child
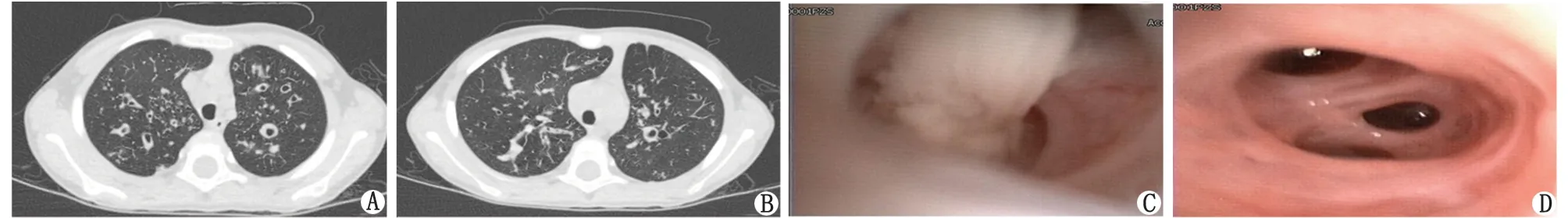
A、B:肺部CT检查,双肺上叶支气管扩张;C:支气管镜检查,管腔内脓性分泌物;D:支气管镜检查,各支气管部分亚段管腔扩张,可见环形皱襞及鱼骨样改变。图2 患儿肺CT及支气管镜结果Fig.2 Lung CT and bronchoscopy results of the child

查见个别纤毛9+2微管结构异常,表现为少数联体纤毛。微管数目正常,内、外动力臂无缺失。图3 纤毛活检电镜结果Fig.3 Electron microscopic results of ciliary biopsy
患儿病史迁延,告知并征得家属同意后,予完善全外显子基因检测。结果显示,CFTR基因异常,检出两个等位基因变异:c.1766+5G>T(Intron 13)剪接突变和c.263T>G(Exon 3)点突变。两个等位基因在CF突变数据库(www.genet.sickkids.on.ca)中均为已知致病性变异,分别来自于患儿父母。结合患儿病史及相关检查,CF诊断明确。制定长期治疗方案:阿奇霉素干混悬剂每次10 mg/kg,1次/d,每周一、三、五口服,3%高渗盐水雾化,补充脂溶性维生素A、D、E、K,乙酰半胱氨酸化痰,孟鲁司特控制气道高反应。长期随诊,加强呼吸道管理,监测生长发育情况。一年来,患儿规则用药,体质量及身高较前增长,偶有轻咳,未因呼吸道感染住院。
1.2 文献复习 在中国知网和万方数据库,以“CF”和“中国人”为关键词进行检索,在PubMed数据库以“cystic fibrosis,Chinese”为检索词,截至2020年12月,共检出华人携带c.1766+5G>T变异10例,携带c.263T>G变异5例,其中1例携带c.1766+5G>T和c.263T>G变异(表1,2)。
携带c.1766+5G>T变异的10例患儿中,男童6例,女童4例;发病年龄1个月~7岁,年龄中位数1岁;诊断年龄1个月~17岁,年龄中位数10岁。值得注意的是,截至目前,该变异仅在中国人群中有报道,而且以南方地区居多,台湾省5例,香港特别行政区2例,厦门市1例。本研究的病例来自泉州市,也属南方地区,可能存在奠基者效应。有文献总结该型基因突变主要分布在东亚[3]。10例患儿均有反复咳嗽咳痰,支气管扩张7例,轻度、中度、重度阻塞性通气功能障碍分别有1、4、1例,鼻窦炎3例,胰腺受累5例,检出铜绿假单胞菌4例,检出金黄色葡萄球菌3例。死亡2例,其中1例为纯合子,发病早,9岁时死亡;另1例合并c.3068T>G变异,23岁时死亡。
携带c.263T>G变异的5例患儿均为女孩,发病年龄3个月~2岁,年龄中位数3个月;诊断年龄4~13岁,年龄中位数10岁,均在北京确诊。5例患儿均有反复咳嗽咳痰、支气管扩张,轻度阻塞性通气功能障碍3例,鼻窦炎2例,变应性支气管肺曲霉菌病2例,胰腺受累3例,检出铜绿假单胞菌4例,检出金黄色葡萄球菌1例。1例纯合子胰腺受累,最终死亡。
携带c.1766+5G>T和c.263T>G变异的1例患儿为女孩,起病年龄2岁,表现为反复咳嗽咳痰、支气管扩张,痰培养出金黄色葡萄球菌[9]。从报道的情况分析,临床表型并不重,暂未发现其他系统受累,经过诊治,仍存活,肺功能提示轻度阻塞性通气功能障碍。本研究报道的病例与该病例相似。
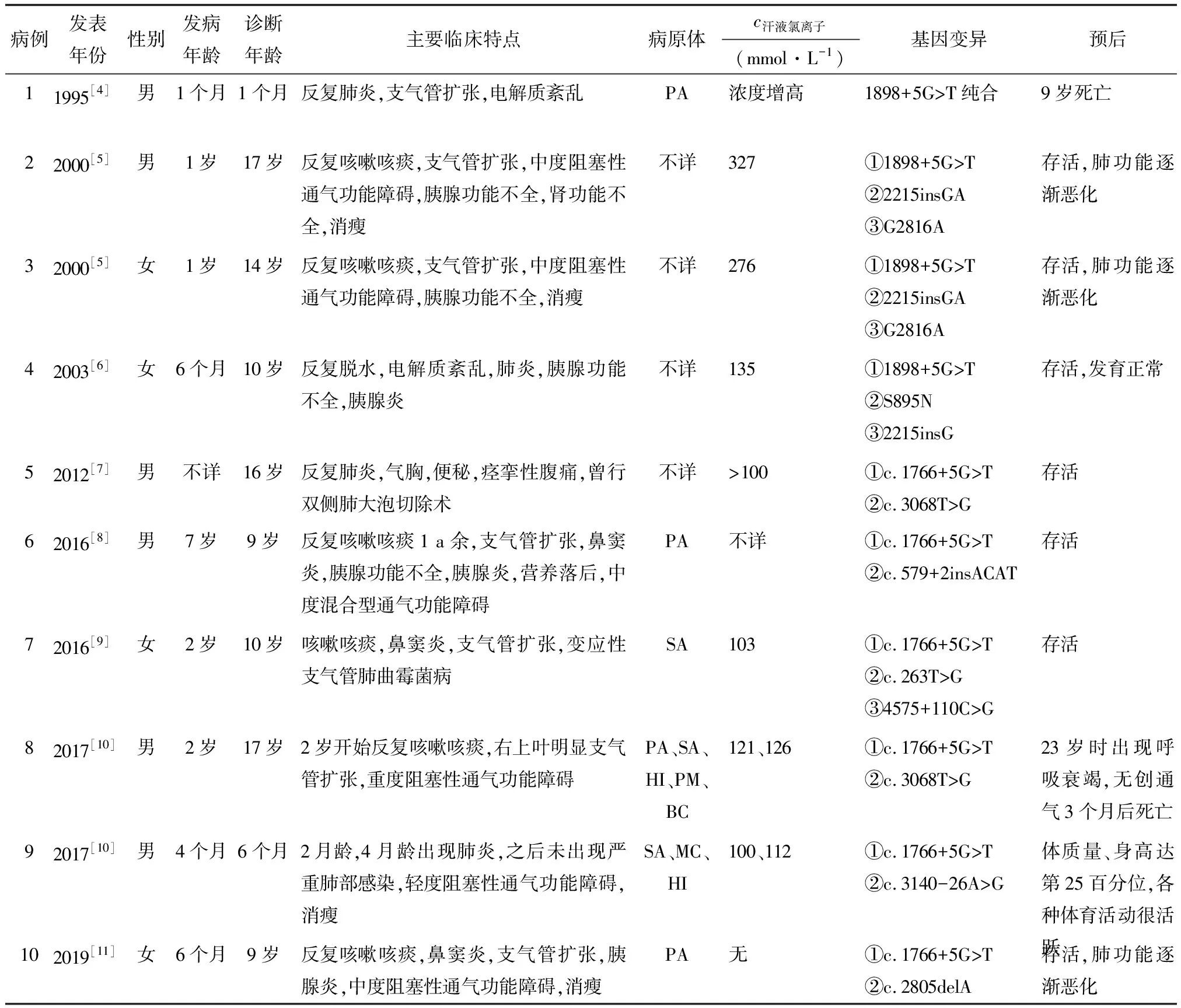
表1 包含c.1766+5G>T位点变异的华人CF患者Tab.1 Chinese CF patients with mutation of c.1766+5G>T

表2 包含c.263T>G位点变异的华人CF患者Tab.2 Chinese CF patients with mutation of c.263T>G
2 讨 论
1949年全球首例CF报道[14]。1989年,Rommens等[15]成功克隆并分离到CF的相关基因,即CFTR存在基因缺陷,位于第7号染色体长臂3区1、2带(7q31.2),全长250 kb,共有27个外显子和26个内含子,cDNA长6 129 bp,编码一条1 480个氨基酸的肽链。CFTR氯离子通道存在于多种细胞膜中(包括汗腺、气道、胰腺、肠道等),主要调节氯通道,使钠吸收增加。功能缺陷的CFTR蛋白通道导致CF患者外分泌功能障碍,氯离子、钠离子、水分子等在CFTR通道的跨膜转运均受到影响,从而产生黏稠分泌物,并在不同器官中堆积、阻塞,形成不同的CF症状。
根据美国CF基金会登记,全世界的CF患者超过70 000例,每年约新增1 000例,>75%的CF患者于2岁前被诊断[16]。目前,我国CF的发病率尚不明确,常被低估,文献报道从发病到最终诊断平均延迟5 a,最长间隔10 a[3]。很多患者早期已有症状,往往辗转多处才得以确诊。目前CF较集中报道于医疗水平发达的地区,我国医务人员对该疾病的认识尚不足。
CFTR基因突变在不同人群中的频率和分布不同。白种人主要突变为F508del,约占70%[16]。中国CF儿童的基因型较分散,以c.1766+5G>T和c.263T>G突变的报道相对较多[16]。一些基因突变在HGMD数据库(http://www.hgmd.cf.ac.uk)中未见报道,如3635delT,c.2907A>C,c.648 G>A,c.960_961insA,c.1075C>T,c.1699G>T,c.3307delA,c.110C>G等,表明CFTR基因存在种族差异[2,9]。
不同的CFTR基因突变与疾病严重程度有关,CFTR基因功能障碍会导致一系列疾病,所涉及的器官数量和疾病严重程度各不相同[17]。CFTR基因突变分为5类[18]。Ⅰ类突变:蛋白质生成缺陷,通常因无义突变、移码突变或剪切位点突变所致,mRNA合成的提前终止和CFTR蛋白的完全缺失。Ⅱ类突变:蛋白质加工缺陷,造成CFTR蛋白翻译后加工异常,从而阻碍蛋白质运输到准确的细胞位置。欧美国家最常见的F508del突变属于此类。Ⅲ类突变:调节缺陷,导致通道对ATP应答的活性降低。Ⅳ类突变:为传导缺陷,蛋白质能合成并准确地定位到细胞表面,但其离子流的速率和通道开放的持续时间减少。Ⅴ类突变:有功能的CFTR蛋白数量减少,包括可改变mRNA稳定性的突变和可改变成熟CFTR蛋白质稳定性的其他突变。一般而言,Ⅰ~Ⅲ类突变引起的疾病较Ⅳ和Ⅴ类严重。c.1766+5G>T变异(曾报道为1898+5G>T)为已知的致病性变异,目前为止仅在中国人群中报道[4-11],其关联的表型主要为严重的肺部疾病。本例患儿等位基因1剪接变异位点c.1766+5G>T位于CFTR基因第12号内含子剪接供体区域,该变异导致mRNA剪接障碍,第12号外显子发生跳跃,导致CFTR蛋白的首个核苷酸结合折叠(nucleotide binding fold,NBF1)区域部分丢失,而该区域与氯离子通道功能相关[9],属于Ⅴ类突变。本例患儿的等位基因2错义突变位点c.263G>T位于CFTR基因第3号外显子第263位碱基为杂合的T>G突变,造成氨基酸p.L88X变异,使第88个氨基酸发生终止,从而影响蛋白的表达[19],属于Ⅰ类突变。CF表型与CFTR基因型相关,还受其他遗传和环境因素的影响[3]。CFTR突变的分类仅作为研究工具,不能预测个别患者的临床结局。对突变情况的了解可能有助于指导初始治疗,但临床应根据观察到的生长情况、肺功能和营养状态进行相应调整。
目前,CF的诊断标准[20]包括:至少存在1个系统CF的典型临床表现;存在CFTR功能缺陷的证据,包括汗液氯离子浓度>60 mmol/L(至少2次),CFTR基因检测显示2个致病性突变位点,鼻黏膜细胞跨膜电位差异常。典型的CF表现为汗液氯离子浓度升高、慢性肺部疾病和胰腺功能不全。Shen等[9]总结了患者的症状体征:反复咳嗽咳痰、支气管扩张、鼻窦炎、痰培养出铜绿假单胞菌是中国CF儿童的重要特征。当患者有反复呼吸道感染、难治性哮喘、变应性支气管肺曲霉菌病时应注意CF,若有胰腺病变、肝胆病变、肠梗阻、巨结肠以及营养不良、严重脱水和电解质紊乱而无明显消化道丢失时,应高度怀疑CF[21]。许多患者症状轻微或不典型,要积极寻找诊断线索。虽然目前国内仅有少数医院可以开展汗液实验,随着二代基因测序技术的开展和普及,CFTR基因变异更容易被发现。由于我国患者的基因型和欧美国家存在较大差异,因此,我国可疑的患者应选择全基因组测序,以便发现少见突变甚至新基因突变。
该病无法治愈,虽然患者的寿命严重缩短,但在过去的60 a,通过支气管扩张剂、抗生素治疗、黏液溶解药物、胰酶替代、加强物理排痰等治疗方案和实施多学科护理,患者的生存率及存活年龄有了显著提高。目前国外的基因治疗效果令人鼓舞[22-26],包括CFTR调节剂,如Ivacaftor(Kalydco)、Lumacaftor/Ivacaftor(Orkambi)、Tezacaftor/Ivacaftor(Symdeko)、Trikafta(tezacaftor+elexacaftor和ivacaftor),但价格昂贵,且任何一种药物都无法治疗存在2 000多个突变类型的疾病[27]。研究中国人群特定的突变类型,为未来CF精准诊断和基因治疗提供可能。
猜你喜欢
保健医苑(2022年1期)2022-08-30
昆明医科大学学报(2022年2期)2022-03-29
中老年保健(2021年11期)2021-08-22
现代临床医学(2021年4期)2021-07-31
支部建设(2020年15期)2020-07-08
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
雕塑(1996年4期)1996-07-12
