
巨噬细胞在肺动脉高压发病中的机制及研究进展
2021-02-12 09:40:48朱宇帆白茜文易舒婷唐瑞娣黄钰航王涛
广州医科大学学报 2021年6期
朱宇帆,白茜文,易舒婷,唐瑞娣,黄钰航,王涛*
(1.广州医科大学第一临床学院,广东广州510180;2.呼吸疾病国家重点实验室广州呼吸健康研究院 广州医科大学附属第一医院,广东 广州510180;3.广州医科大学 广东省血管疾病重点实验室,广东 广州510180)
肺动脉高压(pulmonary hypertension,PH)是由多种病因共同作用引起的一种以肺动脉压力异常升高为特征的病理生理状态,可导致呼吸困难、右心衰竭甚至死亡[1]。血流动学诊断标准为:在海平面、静息状态下,右心导管测量平均肺动脉压≥25 mm Hg(1mm Hg=0.133 kPa)。近期流行病学研究表明,肺动脉高压可能不是罕见疾病,据目前的估计,全球肺动脉高压患病率约1%,其中65 岁以上人群的肺动脉高压患病率高达10%[2]。
按照病因PH 可分为动脉性肺动脉高压、左心疾病所致肺动脉高压、肺部疾病和(或)低氧所致肺动脉高压、慢性血栓栓塞型肺动脉高压和其他肺动脉栓塞性疾病、未明和(或)多因素所致肺动脉高压5 个类别。但它们包含相同的基本病理过程即肺血管重构以及持续的肺动脉收缩,其中以肺动脉内皮细胞(pulmonary artery endothelial cells,PAECs)的功能失调、炎症反应和肺动脉平滑肌细胞(pulmonary arterial smooth muscle cells,PASMCs)的过度增殖等为特征的肺血管重构是PH 的主要病理特点[3]。炎症反应在病理过程中起到了重要作用,其在各种类型的PH 患者和PH 动物模型中都很突出[4]。巨噬细胞(macrophages,mφ)是炎症反应的重要成员:在PH 动物模型以及临床患者的肺组织中,mφ 的密度被发现不仅均明显高于对照的动物或患者[5],且高于左心疾病所致肺动脉高压患者。单核/巨噬细胞谱系是机体防御系统的一个重要组成部分,单核细胞来源于骨髓中的造血干细胞,并在骨髓中发育,当它们从骨髓进入血液时仍然是尚未成熟的细胞,在血液中停留10~20 h 后迁移至组织中,继续发育成mφ。mφ 在肺动脉高压病程中归巢到肺组织后[6],不仅参与了肺血管重构[7],也可以介导肺血管收缩[8]。这些研究结果说明mφ 在PH 的发生与发展中起到了重要作用。
本文旨对mφ 在PH 这一疾病发生发展过程中的作用及其分子机制的研究进展作一综述,系统探讨mφ 在PH 发生发展中的作用,尝试为PH 发病机制的认识以及治疗提供新的思路。
1 肺部mφ 的来源及其在肺组织的归巢
mφ 存在于包括肺脏在内的多种组织器官中,称为组织驻留mφ。肺部的mφ 主要由肺泡mφ 和间质mφ 组成,均参与局部免疫稳态的维持。体内mφ 的来源包括单核细胞起源的mφ、原位增殖而生成的mφ 以及胚胎早期卵黄囊来源的mφ,其中造血干细胞分化产生的单核细胞起源的mφ 是组织驻留mφ 的主要来源。在健康状态下,肺泡mφ 主要来源于胎儿肝单核细胞,间质mφ 则来源于血单核细胞,间质mφ 可转移至肺泡中成为肺泡mφ。当组织发生炎症时,循环中的大量炎性单核细胞被招募到炎症区域并分化成mφ。
通过免疫荧光染色及定量分析,在野百合碱或缺氧诱导的PH 模型中发现mφ 被招募到肺部的血管外膜,且在病变血管周围发现mφ 数量显著增加[8]。PH 大鼠肺组织全转录组测序结果也显示mφ 相关基因显著富集,并且通过针对mφ 标记物甘露糖受体的正电子发射断层扫描发现PH 患者的甘露糖摄取明显高于正常人或左心疾病相关PH 患者[9],这说明mφ 在肺组织的归巢是导致肺血管病变的重要原因,而不是继发于左心疾病导致的肺动脉压力升高的结果。然而,PH 患者肺血管周围归巢的mφ 的来源尚不清楚,有证据表明,肺血管重塑过程中mφ 主要来源于血液单核细胞的归巢和分化[10],但尚未有研究表明没有原位增殖的肺泡mφ的参与。
mφ 在肺组织的归巢会加重PH 肺血管的炎症[11],成为PH 的关键致病因素。CX3CL1/CX3CR系统是单核/巨噬细胞谱系的mφ 在PH 肺组织中归巢的关键信号通路。由HSCs 分化产生的单核细胞分为两个主要亚群:炎性单核细胞(Ly-6chigh,CCR2high/CX3CR1low)和常驻单核细胞(Ly-6clow,CCR2low/CX3CR1high),分别受CCL2 和CX3CL1 两种不同的趋化因子调控。有组织发生炎症时,循环中的大量炎性单核细胞被招募到炎症区域并分化成mφ;然而,在PH 等一些慢性疾病中,常驻单核细胞被证明也具有促炎症特征,CX3CL1/CX3CR1 和CCL2/CCR2 系统均参与PH 病程中单核细胞的募集并相互拮抗[12]。根据Amsellem等的研究[6],CX3CR1 水平升高会导致缺氧诱导的PH 严重程度的加重,伴随肺mφ 数量的增加。这可能是由于在受到刺激时,PAECs 会过度产生趋化因子,如CX3CL1,这触发了表达CX3CR1 的单核细胞对PAECs 的附着从而增加肺单核细胞和mφ 的数量[6],加重肺血管炎症,最终导致PH 的发生发展。
2 mφ 参与的肺血管重构及其机制
肺血管结构细胞(包括PAECs 和PASMCs 等)发生重构是造成PH 的重要原因,会导致肺动脉压力和肺血管阻力的进行性增高。在PH 病程中,mφ归巢到肺组织后,参与了PAECs 的损伤、PASMC 的异常增殖及内膜下迁移、细胞外基质(Extracellular Matrix,ECM)的沉积等肺血管重构过程中的主要事件,从而影响PH 的发生发展。下面将介绍mφ 参与上述各个过程中的主要分子机制。
2.1 mφ 诱导PAEC 的损伤
CD68+单核细胞/巨噬细胞是表达5-脂氧合酶(5-LO)的主要炎性细胞[7],可以通过5-LO 的酶促作用合成白三烯B4(LTB4),参与PH 进程。Tian等[7]报道,在PH 大鼠模型及PH 患者中发现,聚集的mφ 高表达LTB4,通过结合PAECs 上的同源高亲和力G 蛋白偶联受体BLT1,使内皮鞘氨醇激酶1 浓度(Sphk1)减少、活性降低,抑制鞘氨醇-1-磷酸活化内皮一氧化氮合酶(eNOS)[13],减少NO 的产生,氧自由基水平升高,从而直接诱导PAECs 的损伤及凋亡[7],参与了PH 的发生发展。
2.2 mφ 的极化与PASMCs 的增殖和迁徙
mφ 存在两种完全不同的激活状态:M1 样和M2 样表型,M1 样巨噬细胞表现出细胞毒性和促炎表型,具有很强的清除病原体和肿瘤细胞的能力,而M2 样巨噬细胞可抑制免疫和炎症反应,参与组织重塑和肿瘤进展[14]。
在PH 早期,许多研究已发现在肺血管周围有大量mφ 募集。Vergadi 等[10]提出在低氧条件下,肺内mφ 的早期募集和交替激活是缺氧诱导的PH 后期发展的关键:肺泡mφ 在低氧条件下获得交替激活的M2 样表型,通过诱导PASMC 的增殖从而导致血管重塑,促进PH 发展。
在PH 患者中,大部分肺组织细胞都能够释放的IL4 和IL13 这两种主要的M2 表型刺激因子外,也可以通过CD4+T 细胞(包括Th17 细胞)分泌的IL-21 激活下游靶点IL-6 信号[15]或者肺动脉外膜成纤维细胞分泌的IL-6 激活STAT3、缺氧诱导因子1α(HIF1α)和C/EBPβ 信号[16]促进mφ 向M2 表型倾斜,促进mφ 向M2 表型倾斜。
趋化因子CCL2/CCR2 和CCL5/CCR5 系统[17]可能是M2 样mφ 介导PASMC 增殖的重要分子之一。Shariq 等[17]报道,M2 样mφ 通过旁分泌促进PASMCs 有丝分裂,激活后的PASMCs 可以通过旁分泌的方式促进mφ 极化为M2 样表型,mφ 和PASMCs 共同表达的趋化因子受体CCR2 及CCR5可能是这种协同机制的基础[17]。这表明巨噬细胞在PASMCs 附近活化成M2 表型可能会导致恶性循环,使mφ 和PASMCs 相互激活并放大彼此的相互作用,最终对PASMCs 增殖发挥重要的作用。趋化因子受体CX3CR1 也可能是M2 样mφ 诱导PASMC增殖的重要分子之一,一系列体外和体内实验[6]表明抑制M2 样mφ 表达的CX3CR1 可以减缓PASMCs 增殖,延缓甚至是逆转肺血管重构。
此外,slug 转录因子及其靶蛋白催乳素诱导蛋白(prolactin induced protein,PIP)也可能参与了mφ诱导的PASMCs 增殖。有研究发现[18],肺纤维化相关的肺动脉高压(pulmonary fibrosis-pulmonary hipertension,PF-PH)患者肺纤维化和非纤维化区域的血管壁厚度比肺纤维化(pulmonary fibrosis,PF)患者显著增加,这与mφ 在PF-PH 患者肺血管中的作用有关:Slug 转录因子在肺上皮细胞、mφ 和成纤维细胞中均有表达,但在PF-PH 中只有mφ 的Slug表达明显高于PF 患者,PIP 水平上调[18],诱导PASMCs 增殖;同时,M2 样mφ 可以表达高亲和力的IL13Rα2 上调转化生长因子-β1(transforming growth factor-β1,TGF-β1)表达,从而促进肺血管周围纤维化,加重血管壁增厚,增加肺血管阻力,导致PH 的发生发展[19]。
2.3 mφ 促进ECM 在肺动脉的沉积
ECM 的沉积是PH 病变肺血管重塑过程中主要的事件之一,可导致血管壁增厚和管腔狭窄[8]。mφ 可通过表达天冬酰胺内肽酶(legumain,LGMN)促进ECM 的合成,LGMN 是新发现的属于C13 肽酶家族的半胱氨酸蛋白酶[20],主要在mφ 中表达。在肺动脉中,LGMN 可以通过激活基质金属蛋白酶-2将TGF-β1 前体水解为活性形式,促进ECM 合成[21],增加肺动脉管壁厚度。临床上,血清LGMN水平与特发性肺动脉高压的严重程度密切相关。
3 mφ 参与肺动脉收缩
Kumar Rahul 等[22]报道,在低氧条件下,小鼠的单核细胞被招募为间质mφ,表达血小板反应蛋白-1(Thrombospondin-1,TSP-1),从而导致TGF-β 活化,激活下游靶点Rho 激酶,介导血管收缩。缺氧诱导的PH 小鼠与正常小鼠相比,前者的间质mφ 中编码TSP-1 的基因Thbs1 的表达较高[23],间质mφ 在缺氧条件下从血管腔被招募到肺组织并分泌TSP-1,在TSP-1 介导的TGF-β 通路激活下,Rho 激酶活性升高,从而抑制肌球蛋白磷酸酶的磷酸化,促进肌球蛋白的活性,引起肺血管收缩。
4 针对mφ 在肺动脉高压中的治疗策略
目前针对mφ 治疗肺动脉高压的研究发展迅速,涉及多个方面,并在细胞实验及动物模型中都证实了较好的疗效,有望成为未来肺动脉高压治疗的关键靶点。
如前文提到,CX3CL1/CX3CR 系统是PH 肺组织中mφ 归巢、mφ 介导PASMCs 增殖的关键信号通路,通过基因或药物抑制CX3CR1 可延缓PH 的发生发展[6]。
M2 样mφ 可以促进PASMCs 的增殖,可以针对这一效应制定治疗策略,包括降低逆转M2 样mφ 的转化和降低M2 样mφ 存活率,如(1)通过调节微环境降低M2 样mφ 基因的表达[24];(2)静脉注射间充质细胞来源的外泌体可阻碍缺氧状态下各种信号转导,抑制与mφ 交替激活关系密切的STAT3 的激活和miR-17 超家族microRNA 簇的上调,最终减少炎症及PASMCs 增殖的发生[25];(3)通过IL-6/IL-21 信号轴阻断mφ 向M2 样倾斜,在上游用抗IL-6受体的单克隆抗体MR16-1 阻断IL-6,从而阻止缺氧性Th17 细胞和M2 样mφ 在肺内的聚集,从而延缓缺氧性PH 的发生发展[15]。
另一方面,通过阻断LTA4H 降低mφ 表达的LTB4 水平,恢复Sphk1-eNOS 信号转导,可以阻止PAECs 凋亡,逆转PH[7]。此外,也有研究发现特异性抑制与ECM 沉积密切相关的LGMN 可明显改善PH[8]。
综上所述,以肺血管周围mφ 的浸润为特征的炎症反应是PH 的关键致病因素。mφ 在肺动脉高压病程中归巢到肺组织,不仅参与肺血管重构,也可以介导肺血管收缩(见图1)。以mφ 为靶点治疗PH 的在细胞实验及动物模型中都证实了较好的疗效并取得了重要突破,有望成为未来肺动脉高压治疗的新靶点。
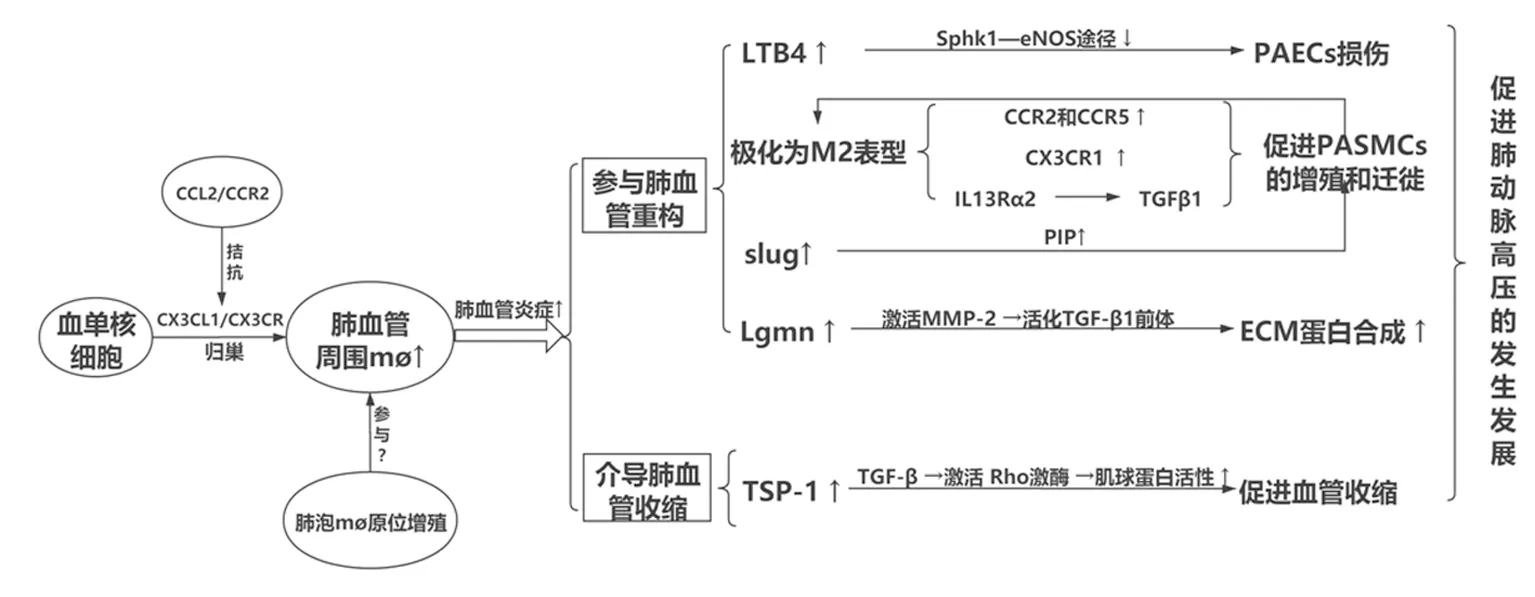
图1 巨噬细胞在肺动脉高压发病中的作用
猜你喜欢
小学生学习指导(低年级)(2023年6期)2023-06-30 07:49:10
大理文化(2020年3期)2020-06-11 00:41:51
现代园艺(2017年21期)2018-01-03 06:41:32
军营文化天地(2016年10期)2016-06-15 20:28:30
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
温州医科大学学报(2014年7期)2014-07-18 02:43:14
现代检验医学杂志(2014年5期)2014-02-02 02:51:34
食品科学(2013年15期)2013-03-11 18:25:40
