
克氏综合征
2021-02-11 02:43赵馨纪媛君王秋明骆玉梅陈耀勇胡芷洋许曼肖建平
中国产前诊断杂志(电子版) 2021年4期
赵馨 纪媛君 王秋明 骆玉梅 陈耀勇 胡芷洋 许曼 肖建平*
(1.无锡市妇幼保健院医学遗传与产前诊断科,江苏无锡 214002;2.中山大学附属第一医院妇产科,广东广州 510080;3.广州医科大学附属第三医院广东省产科重大疾病重点实验室、广东省高校生殖与遗传重点实验室,广东广州 510150;4.深圳市人民医院产前诊断中心,广东深圳 518020)
克氏综合征(Klinefelter syndrome,KS)是指男性中存在多余的X染色体导致的疾病,核型一般表现为47,XXY,是最常见的性染色体非整倍体,新生儿中的发生率约为1/660[1,2]。考虑到表型轻微的病例可能未得到诊断,其发生率可能被低估。贯穿KS患者一生的特征性症状主要体现在生理和神经发育层面,包括生长、认知发育、内分泌和生殖,异常表现包括高身材、性腺功能减退、男性乳房肥大症、不育、代谢异常等[3]。克氏综合征具有极强的临床异质性,患者可能有不同程度的认知、社交、行为和学习障碍[4]。在一些特殊的病例中可能存在不止1条多余的X染色体,如48,XXXY和49,XXXXY,其发生非常罕见,这些患者除了有47,XXY患者的临床特征外,通常还具有更严重的智力障碍和骨骼异常等问题[5]。KS的遗传咨询倾向于重点关注该疾病的远期预后,包括生长发育、智力情况和健康状况等[3,6]。
1 临床特征
KS男性通常睾丸小、睾酮水平低、促性腺激素水平高;部分患者生殖器官异常,包括睾丸未下降(隐睾症)、尿道下裂、小阴茎等[7]。KS患者睾丸间质细胞增生,不能生成充足的睾酮[8]。在得不到治疗的情况下,睾酮的不足将导致患儿青春期延迟或者发育不完全,男性乳房肥大,肌肉含量下降,骨密度下降,头发和体表毛发稀疏等[9]。研究发现大多数KS患儿青春期发育正常,然而其睾丸体积不超过4~5ml,第二性征出现平均晚3~4年[10]。50%~75%的KS男性乳腺组织增多,其中20%的患者可表现为男性乳房肥大症,这可能是由于芳香化酶CYP19的过表达导致[11]。KS男性睾丸中原始生殖细胞退化迅速,青春期时睾丸中几乎不存在原始生殖细胞[10,12]。成年的KS男性生精小管广泛纤维化和透明化,生成的精子数量非常少,超过90%的病例表现为无精子症或不育[13]。
KS患者出现社交障碍、焦虑、抑郁和行为问题的风险增加[14],大约10%的患者合并自闭症谱系障碍[15]。随着大脑神经成像技术的快速发展,KS患者的神经发育特征谱被进一步扩大与定义[16]。KS患儿的神经成像研究发现其大脑尾部、额叶和颞叶区域的容量减小。磁共振显示KS患儿大脑尾部和前脑的异常与多动症儿童相似[16]。未经干预治疗的KS患者大脑颞叶区受损,可能导致阅读障碍、语言信息处理障碍和社交障碍等[17]。虽然KS患儿通常不存在明显的智力障碍,但是会表现为交流和表达的障碍,通常语言理解能力较语言表达能力强[18]。KS患者大脑岛叶区灰质密度降低,可能与情绪情感处理能力缺陷有关。一项研究显示,4~9月龄期间启动早期激素替代治疗的KS患儿,其大脑神经系统发育可接近正常[19]。
KS男性的身高普遍高于同龄人,成年后身高可达到第75~90百分位[20]。其高身材可能是由于性染色体上的身高决定基因SHOX的过表达[21],也有学者认为雄激素水平不足导致的骨骺融合延迟也可能导致KS患儿的高身材[22]。少部分KS患儿可能会有不同程度的肌肉骨骼异常表现,包括脊柱侧弯、斜颈、驼背、扁平足、韧带松弛、桡尺骨骨性联接、第五指弯曲和漏斗胸等,其中一些异常表型通常不易察觉[23,24]。
获得临床诊断的KS男性平均生活质量低于同龄健康人群,预期寿命平均缩短约一年半至两年[25]。尽管相关的纵向研究数据有限,一些研究认为KS男性患心血管和代谢异常相关疾病的风险增加[26]。相较于健康男性,KS患者发生震颤、骨质疏松、自身免疫性疾病如系统性红斑狼疮、类风湿性关节炎和干燥综合征的风险增加[27,28]。同时KS男性患乳腺癌、非霍奇金淋巴瘤与肺癌的风险也增加[27]。
核型为46,XY/47,XXY嵌合体的KS患者临床表现和症状通常难以预估,取决于哪些组织或器官的细胞中存在多余的X染色体。与典型的KS患者(47,XXY)相比,核型为48,XXXY和49,XXXXY的KS患者临床表现一般更严重,主要包括学习困难、智力障碍、语言发育迟缓、肌张力低下、性腺功能减退、生长发育迟缓、特殊面容(眼距过宽、上睑裂、内眦赘皮等)、生殖和肌肉骨骼系统异常等[5],其中大多数患者的男性性腺发育受严重影响,睾丸通常无精子,部分患者有比较严重的智力障碍[29]。
2 遗传学诊断
染色体核型分析和染色体微阵列分析等检测技术可以诊断KS[7],KS的早期诊断被证实有助于监测和评估患者未来可能出现的发育异常[30]。1项对2岁以下的非嵌合型KS患儿的研究显示,大多数患儿一般是由于出现大运动和(或)语言发育的落后行遗传检测得到诊断[19]。KS患儿婴儿期即获得诊断通常是由于已出现明显的生理异常,包括尿道下裂、小阴茎或者隐睾等[31]。KS男性性腺功能减退的症状通常青春期中期开始显现,直至成年早期表现明显,并且随着年龄增长逐步进展。临床医生通常会建议青春期延迟、小睾丸或疑似不育的男性行遗传学检测以排除KS。研究显示11%的无精子症男性和4%的不育男性可能为KS患者[32]。KS患者父母如果染色体核型正常,其再次生育KS患儿的风险与普通人群类似[7]。目前大概只有10%的KS病例在产前得到遗传学诊断[33]。无创产前检测(non-invasive prenatal testing,NIPT)技术的应用使得产前发现KS胎儿的概率增加[34]。一些主流的学术团体已推出专家共识支持使用NIPT技术检测性染色体非整倍体,包括KS[35,36]。需要注意的是,NIPT技术不是诊断技术,阳性的NIPT结果需要通过侵入性产前诊断技术得到确认[37]。
本文章纳入并统计了4家医院(无锡市妇幼保健院、中山大学附属第一医院、深圳市人民医院、广州医科大学附属第三医院)不同来源样本(产后外周血、流产组织和产前诊断)中遗传诊断为KS相关的病例,包括47,XXY、47,XXY嵌合体、48,XXXY、48,XXYY和49,XXXXY的病例(表1)。产后外周血样本中47,XXY和47,XXY嵌合体发生率分别为1/147和1/3322;流产组织样本中47,XXY和47,XXY嵌合体发生率分别为1/1256和1/942;产前诊断样本(包括羊水、绒毛和脐带血)中47,XXY和47,XXY嵌合体发生率分别为1/335和1/2406。产后外周血样本中47,XXY的发生率较流产组织样本和产前诊断样本高,这可能是源于3个样本组受检病例的临床表现不同,可能存在不可避免的选择偏倚。不同来源的样本中48,XXXY、48,XXYY和49,XXXXY病例均非常罕见(表1)。

表1 不同来源样本中KS相关病例的发生率
产前诊断为KS后选择终止妊娠的病例约占11.6%~87.5%,这一比例在不同的国家和地区有所不同[3]。这可能与公众对KS这种疾病的认识有关,也可能与当地传统、宗教信仰和合法性等有关。我们评估了2家医院(无锡市妇幼保健院、中山大学附属第一医院)产前诊断为KS相关病例的产前诊断指征以及妊娠结局,产前诊断总例数为28 230例。产前诊断为47,XXY和47,XXY嵌合体的病例中产前诊断指征主要为NIPT高风险、高龄和血清学筛查高风险(表2)。产前诊断为47,XXY的病例中,选择引产的病例占63.0%,选择继续妊娠(早产或足月产)的病例占13.7%,失访的病例占23.3%;产前诊断为47,XXY嵌合体的病例中,选择引产的病例占30.8%,选择继续妊娠的病例占46.2%,失访的病例占23.1%(表3)。

表2 产前诊断为KS相关病例的产前诊断指征(例)
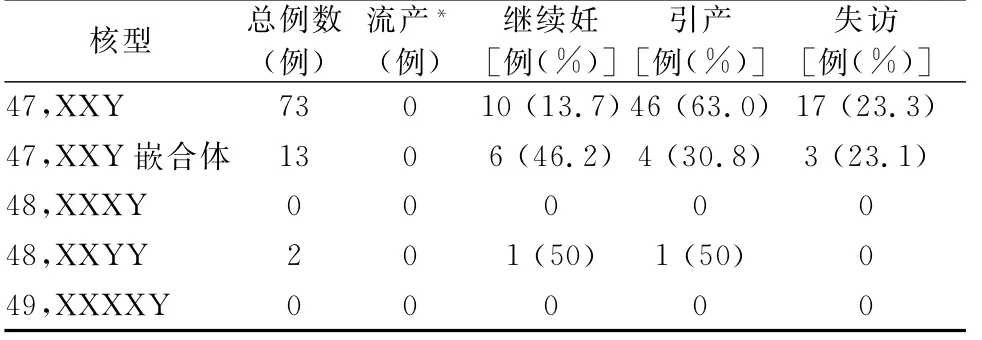
表3 产前诊断为KS相关病例的妊娠结局
美国一项回顾性队列研究发现,产前诊断为KS的病例不良妊娠结局的风险增加,其中早产、剖宫产、新生儿呼吸窘迫综合征和小于胎龄儿的概率显著增加[38]。这一结果提示我们要对产前诊断为KS后选择继续妊娠的病例密切随访,监测其发生早产和新生儿不良结局的风险。
3 发生机制
3.1 47 ,XXY 90%以上的KS患者核型为纯合型47,XXY。47,XXY通常是由于生殖细胞形成过程中随机发生的性染色体不分离导致生殖细胞中X染色体数目异常[39]。多余的1条X染色体源自父亲和母亲的概率大约各为50%[40]。目前没有证据显示47,XXY中多余的X染色体的亲本来源对患者表型有显著影响。高龄母亲(≥35岁)生育47,XXY后代的风险增加[41]。母亲高龄被认为与卵母细胞第一次减数分裂错误有关[42]。与小于24岁的女性相比,40岁以上女性孕育47,XXY胎儿的风险增加了4倍。小于10%的KS患者为46,XY/47,XXY嵌合体,其发生是由于胚胎发育早期细胞分裂过程中X染色体不分离[7]。
3.2 48 ,XXXY和48,XXYY 48,XXXY通常是由于生殖细胞(精子或卵子)形成过程中连续发生的2次性染色体不分离事件导致生殖细胞中存在多余的2条X染色体[5]。48,XXYY通常是由正常的卵子(Xm)和异常精子(XpYpYp)受精形成,该异常精子(XpYpYp)的产生是由于精母细胞连续的2次减数分裂过程中性染色体不分离[43]。已报道的文献中共有11例48,XXYY病例接受了性染色体亲本来源检测,结果显示所有病例中多余的X和Y染色体均为父本来源[5]。
3.3 49 ,XXXXY 49,XXXXY通常是源于卵母细胞第一次减数分裂和第二次减数分裂过程中的X染色体不分离导致卵子中存在4条X染色体,即为异常卵子(XmXmXmXm)和正常精子(Yp)受精形成的个体[44]。
许多研究尝试去寻找KS患者神经发育与行为异常轻重程度可能的遗传学机制。目前已知人类X染色体包含842个编码基因和629个非编码基因(Ensembl release 87,December 2016)。X染色体上约15%的基因可以逃避X失活[45]。有研究认为KS患者的异常表型可能主要源于多余的X染色体上逃避失活基因的剂量效应干扰了正常的发育过程,包括胎儿期和青春期的生殖系统发育以及出现其他可能的症状[46],然而影响到最终表型的基因数量和基因名称目前尚无定论[47]。另外有学者认为KS患者临床表型的异质性可能与X失活偏移、嵌合情况以及表观遗传修饰等有关[29]。
4 治疗与预后
KS患者的干预治疗通常是基于其临床表现和症状[7]。在婴儿期和学前阶段,KS患者常常面临生长发育、运动、语言和行为发育的缺陷;进入学习阶段后,他们主要面临语言表达障碍、执行功能障碍、学习障碍等困难[14]。多种形式的疗法,包括物理、语言、行为、心理和家庭疗法,通常可以帮助减轻KS患儿的部分症状,帮助他们在学校更好地表现[46]。早期以及持续的干预、制定教育计划和社交技能训练对于KS患儿发展技能至关重要[5]。有乳房增大表现的KS患者,可以通过包括雌激素抑制剂在内的药物干预治疗,诊断为男性乳房肥大症的患者可通过乳房缩小或切除手术治疗[11]。
睾酮水平低下的KS患者可以通过外源补充睾酮[48]。维持正常的睾酮水平可以改善患者的肌肉发育、嗓音、毛发;提高患者的性欲、增大睾丸体积;改善患者的情绪、自信力和行为;预防骨质疏松症并降低患者自身免疫性疾病和乳腺癌发病的风险[46]。研究显示,KS患儿在青春期中后期开始进行睾酮替代治疗可以使青春期发育完全,维持与年龄相匹配的男性第二性征[49]。但是目前为止,仍然缺乏科学的临床对照试验提供充分证据支持KS患儿青春期前启动雄激素替代治疗的安全性和有效性,睾酮的补充剂量也没有标准化[13]。未来需要大样本的前瞻性队列研究和长期的随机对照研究,关注不同年龄阶段起始激素替代治疗对KS患儿健康的影响。
成年的KS男性通常生育力低下[7]。近年来得益于辅助生殖技术的发展,KS男性可通过睾丸穿刺取精和卵胞浆内单精子显微注射技术生育后代[50,51]。2016年一项临床研究显示,16~30岁的KS男性通过睾丸穿刺获取精子的成功率为40%~70%[52];精子获取后的成功受精率接近50%[53]。一项胚胎植入前遗传学诊断发现,KS男性后代胚胎性染色体非整倍体的概率约为该中心胚胎植入前遗传学检测(preimplantationgenetictesting,PGT)周期平均值的2倍[54]。因此,KS男性的伴侣妊娠后需行产前遗传学诊断。
5 结论与展望
过去的10年中,由于遗传学诊断技术的发展,KS的诊断率逐步提升。KS患者贯穿生命周期的持续的发育评估、心理评估和干预治疗,需要多学科专家团队的参与,包括儿科、泌尿外科、内分泌科、语言康复、心理咨询和生殖医学等。及时诊断,早期干预和多学科团队的健康管理对于KS患者的神经系统发育、生命健康和生育力有重要意义。
猜你喜欢
人人健康(2022年18期)2022-10-10
现代医学与健康研究电子杂志(2021年21期)2021-12-26
祝您健康·文摘版(2019年1期)2019-05-14
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
家庭用药(2016年9期)2016-12-03
中学生理科应试(2016年4期)2016-11-19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
现代家庭(1999年7期)1999-06-14
祝您健康(1991年1期)1991-12-30
