
Changes in gut microbiota composition and diversity associated with post-cholecystectomy diarrhea
2021-02-05 03:36YanDongLiBaoNingLiuSiHaiZhaoYongLiZhouLiangBaiEnQiLiu
Yan-Dong Li, Bao-Ning Liu, Si-Hai Zhao, Yong-Li Zhou, Liang Bai, En-Qi Liu
Abstract
Key Words: Cholecystectomy; Post-cholecystectomy; Diarrhea; 16S rRNA; Microbiota; Bifidobacterium
INTRODUCTION
Cholecystectomy, surgical removal of the gallbladder, can alter the flow of bile to the intestines. There are approximately 1013bacteria in the human intestines, which can be divided into three categories according to their functions: Beneficial, harmful, and neutral bacteria[1].
After cholecystectomy, bile enters the duodenum directly, independent of the timing of meals. The bile acid metabolism balance is disrupted and primary bile acid production is increased. The interaction between the bile acids and the intestinal microbes is changed[2]. The occurrence of post-cholecystectomy diarrhea (PCD) may be related to the change in the bile flowing into the intestines after cholecystectomy[3,4]. Some studies have provided evidence that the intestinal microbiota maintains normal gastrointestinal function and that alterations in the intestinal microbial community contributes to gastrointestinal symptoms[5-9]. Accumulating evidence indicates that the gut microbiota plays a significant role in the development of a number of disease states, such as immune, metabolic, and even skin diseases[10-13]. Studies have shown that gut dysbiosis may also cause many diseases, such as diarrhea, inflammatory bowel disease, obesity, and diabetes[14-16].
Thus, PCD may be related to the gut microbiota. PCD is part of the postcholecystectomy (PC) syndrome, and is difficult to treat[17]. The clinical characteristics of PCD include feeling an urgent need to defecate, an increased frequency of defecation, and diarrhea that mostly occurs in the daytime. According to the definition of diarrhea as three or more bowel movements of liquid stools per day without precipitating factors[18,19], our questionnaire survey of PC patients showed that diarrhea occurred in 32% of patients after 6 mo (data not shown). Studies have reported that cholecystectomy is associated with a higher occurrence of diarrhea[20]. Approximately 12%-35.6% of PC patients have varying degrees of chronic diarrhea[21-25].
Little is known regarding the intestinal microbiota characteristics in PCD patients. To better understand the role of the intestinal microbiota in PCD patients, highthroughput sequencing of the 16S rRNA gene was used to characterize the composition and diversity of the complex intestinal microbial community for the first time. In this study, we identified changes in the fecal microbiota in PCD patients. The results indicated that decreased diversity and abundance of the microbial community in the PCD group may cause diarrhea.
MATERIALS AND METHODS
Study population
We studied 31 PC patients who were divided into two subgroups: The PCD group (n= 16) and the post-cholecystectomy non-diarrhea (PCND) group (n= 15). A group of 20 healthy controls (HC) was also included, who had a similar age range and sex ratio as the PC group (Table 1). Subjects were recruited from the First Affiliated Hospital of Xi’an Medical University, China, and health status was assessed at the participants’ annual physical examination appointments. The inclusion criteria for PC patients were as follows: No retained gallstones, no bile duct injury, no stenosis of the bile duct, and ≤ 1 year since cholecystectomy. The exclusion criteria for PC patients were as follows: Alcohol consumption, smoking, history of treatment with antibiotics or antiinflammatory agents in the previous 3 mo, history of other diseases in the previous 3 mo, and consumption of probiotics in the previous 2 mo.
Ethics statement
The study was undertaken with the approval of the Medical Ethics Board of the First Affiliated Hospital of Xi’an Medical University. All experiments were performed in accordance with the approved guidelines. All participants provided written informed consent before participation in the study.
Sample collection and DNA extraction
Fresh stool samples from each participant were collected in the morning using fecal collection containers. Each sample was immediately transferred to the laboratory where it was homogenized, divided into aliquots, and stored at -80℃ before further analysis. Bacterial DNA was extracted from the stool samples using a QIAamp DNA Stool Minikit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
16S rRNA amplicon sequencing
The 16S rRNA gene (V3-V4 region) in the extracted DNA was amplified using the primer pair F (5’-ACTCCTACGGGRSGCAGCAG-3’) and R (5’-GGACTACV VGGGTATCTAATC-3’). A KAPA HiFi Hotstart ReadyMix PCR kit was used for the high-fidelity amplification. The rDNA was then sequenced using an Illumina HiSeq 2500 platform (RealgeneBio Co. Ltd., Shanghai, China) with 2 × 250 base pair pairedend sequencing. The read length was centralized on 400-440 base pairs. Raw data are now available at NCBI under the Sequence Read Archive database with accession no. SRP247004.
Sequencing data analysis and statistical analysis
PANDAseq was applied to assemble the overlapping paired-end reads[26]. The 16S rDNA sequences were clustered into operational taxonomic units (OTUs) based on ≥ 97% sequence similarity using USEARCH[27]. A representative sequence for each OTU was chosen for downstream analysis based on the most abundant sequence. OTUs were taxonomically assigned using the Ribosomal Database Project (RDP) database and classifier (RDP, http://rdp.cme.msu.edu)[28,29].
Quantitative Insights Into Microbial Ecology was used to generate the rarefaction curves for testing the sequencing depth. Phylogenetic diversity tree diversity indices
were used to assess the α-diversity, which represents the species abundance in a single sample, and comparisons between groups were performed. The communities were compared based on phylogenetic distances using the weighted UniFrac metric to assess β-diversity. One-way analysis of similarities (ANOSIM), nonmetric multidimensional scaling (NMDS), and multi-response permutation procedure (MRPP) analyses were also employed to assess the community similarities or dissimilarities between groups. Linear discriminant analysis (LDA) effect size (LefSe) was applied to identify bacteria with differential abundance between groups. LefSe analysis was applied with a logarithmic LDA score cut off of 2. Statistical analysis of microbial differences between groups was performed using the Wilcoxon rank sum test and Kruskal-Wallis rank sum test in R3.1.0. All values ofP< 0.05 were considered significant. Spearman’s correlation coefficient was calculated using R and visualized as a network using corrplot.
Analysis of predicted metagenomes
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used for analyzing both 16S rRNA gene relative abundances and the predicted metabolic data. The sequences of genes in the merged gene catalogue were aligned to the Kyoto Encyclopedia of Genes and Genomes (KEGG) bioinformatics database.
RESULTS
Diversity and richness of gut microbiota decreased after cholecystectomy
The basic information on the study population is listed in Table 1. The diversity and profiles of the gut microbiota were analyzed by performing high-through put 16S rRNA gene sequencing in the PC and HC groups. In total, 720 OTUs were identified based on a 3% dissimilarity cut-off. To better understand the differences in the microbiota distributions between groups, a Venn diagram was constructed and it was found that the majority of OTUs were shared by the HC and PC groups. However, the PC group had fewer OTUs than the HC group (Figure 1A). We also observed attenuated α-diversity in terms of community richness in the PC group, although the difference was not statistically significant (Figure 1B). Based on the β-diversity indices of the gut microbiota in the PC and HC groups, NMDS was implemented to assess discrepancies regarding different abundances, which indicated that β-diversity was decreased in the PC group (Supplementary Figure 1). Furthermore, the microbiota abundance, from phyla to genera, was different between the PC and HC groups. Based on profiling of bacterial taxa, the abundance of the genus Prevotella was higher and Bacteroides was lower in the PC group compared to the HC group (Figure 1C). There were 20 bacterial taxa (seven at the genus level) that exhibited remarkable differences between the HC and PC groups. At the genus level, Bifidobacterium, Acinetobacter, and Megasphaera were higher in the PC group, while Clostridium XVIII, Brachybacterium, Leucobacter, and Clostridium sensu stricto were higher in the HC group (Supplementary Figure 2). Using LefSe analysis, 15 taxa with differential abundance between the HC and PC groups were identified (Figure 1D).
Diarrhea-associated intestinal microbiota in PC patients
By comparing the OTUs between the PCD and PCND groups, we found attenuated community richness in the PCD group. It was found that 427 OTUs were shared between the PCD and PCND groups, the PCND group had 75 OTUs more than the PCD group. Significant differences in β-diversity between the PCD and PCND groups were found based on the weighted UniFrac metric (ANOSIMr= 0.137,P= 0.006) (Figure 2A), indicating that the fecal microbial structure and richness in the PCD group were significantly different compared to the PCND group. MRPP analysis also indicated a significant difference between the PCD and PCND groups based on the weighted UniFrac metric (P= 0.009). Based on the NMDS plot, the patients formed two clusters: A tight PCD cluster (Figure 2A, green dots) and a tight PCND cluster (Figure 2A, yellow dots).
Profiling of the bacterial taxa was conducted at the phylum and genus levels. At the phylum level, the abundance of Bacteroidetes was significantly higher in the PCD group than in the PCND group. The ratio of Firmicutes/Bacteroidetes was decreased in the PCD group compared with the PCND group (Figure 2B). However, at the genus level, Bacteroides decreased in the PCD group compared with the PCND group (Supplementary Figure 3). LefSe analysis revealed that 23 bacterial taxa (with nine at the genus level) exhibited remarkable differences between the PCD and PCND groups (Figure 2C). The Wilcoxon and Kruskal-Wallis rank sum tests identified the top 20 differentially abundant microbiota between the PCD and PCND groups, and the relevant eight taxa at the genus level are shown in Figure 2D. At the genus level, Prevotella and Sutterella were higher in the PCD group, while Bifidobacterium, Citrobacter, Clostridium IV, Clostridium XlVb, Lactococcus, and Raoultella were higher in the PCND group (Figure 2D).

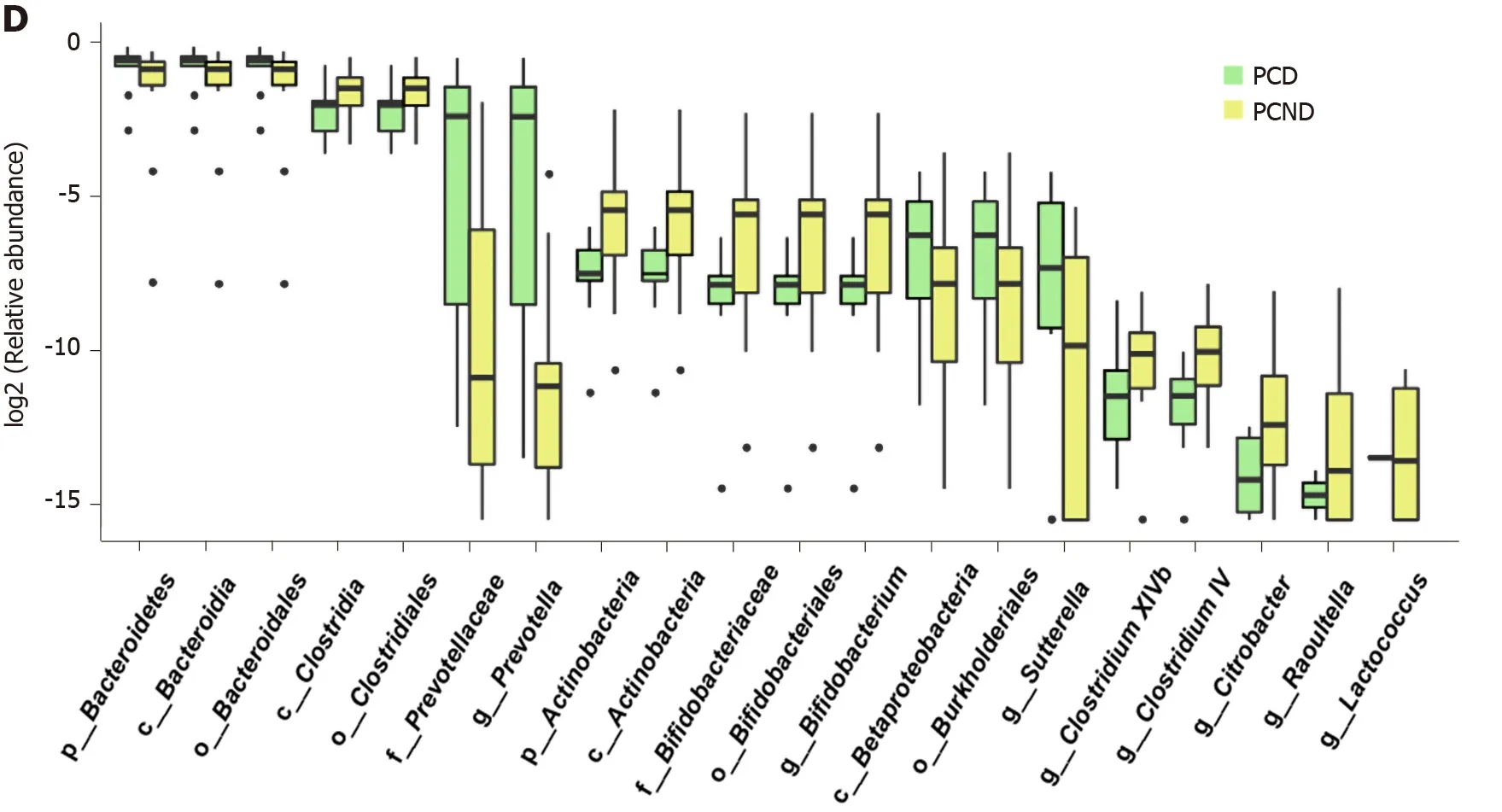
Figure 2 Comparison of gut microbiota structure and abundance between the post-cholecystectomy diarrhea and post-cholecystectomy non-diarrhea groups. A: Differences between and within post-cholecystectomy diarrhea (PCD) and post-cholecystectomy non-diarrhea (PCND) groups were assessed by one-way analysis of similarities and nonmetric multidimensional scaling analysis of the gut microbiota based on the weighted UniFrac metric; B: Comparison of the relative abundance at the phylum level between the PCD and PCND groups; C: Linear discriminant analysis effect size analysis showing the taxa with the greatest differences in abundance between the PCD and PCND groups. Green bars: PCD group-enriched taxa; yellow bars: PCND group-enriched taxa. The brightness of each dot is proportional to the effect size; D: Box plots showing the relative abundance of the top 20 microbial taxa with differential abundance. PCD: Post-cholecystectomy diarrhea; PCND: Post-cholecystectomy non-diarrhea; LDA: Linear discriminant analysis.
Correlations among diarrhea-associated microbes
The differentially abundant microbiota could be used to clearly distinguish the PCD group from the PCND group (Figure 3A). To further characterize the relationships between the bacterial taxa, we calculated the Spearman’s correlations of the bacterial genera with differential abundances (Figure 3B). Significant positive correlations were observed among Lactococcus, Citrobacter, and Raoultella, while a significant negative correlation was observed between Prevotella and Bifidobacterium.
Predictive function analysis of bacterial taxa associated with diarrhea
The KEGG analysis suggested that the fecal microbial structures may contribute to functional variation in their hosts. At KEGG levels 2 (Figure 4A) and 3 (Figure 4B), we identified seven and 33 differentially abundant pathways, respectively. At level 2, seven microbial gene functions, including pathways involved with amino acid metabolism, lipid metabolism, and the circulatory system, were higher in the PCND group, whereas microbial gene functions related to the digestive system, metabolism of terpenoids and polyketides, folding, sorting, and degradation, and energy metabolism pathways were higher in the PCD group.
DISCUSSION
Cholecystectomy has become the most common biliary surgery[30], but the main complication of diarrhea has been largely ignored. It affects the quality of life of PC patients to a certain extent[31,32]. Therefore, it is necessary to pay attention to PCD.
In this study, we mainly analyzed and compared the fecal microbiota communities between PCD and PCND patients using 16S rRNA gene sequencing. Our study found a significant reduction in α-diversity in the fecal microbiota of PCD patients, which suggests that the fecal microbiota of PCD patients has lower microbial richness and diversity.
To compare the different fecal microbial structures between groups, we calculated the UniFrac phylogenetic distances related to the microbe composition among subjects. NMDS analysis was used to distinguish the clustering of the PCD group from the PCND group, and distinct bacterial profiles were observed. The β-diversity index (structure) in the PCD group differed significantly from that in the PCND group, indicating a significant difference in bacterial diversity between the PCD and PCND groups.

Figure 3 Principal component analyses and correlation plots of different microbial taxa in the post-cholecystectomy diarrhea and postcholecystectomy non-diarrhea groups. A: Principal component analysis plot of different taxa in the post-cholecystectomy diarrhea and post-cholecystectomy non-diarrhea groups; B: Spearman’s correlation plots of the relative abundances of the differentially abundant bacteria at the genus level. Statistical significance was determined for all pairwise comparisons. Positive values (blue circles) indicate positive correlations, and negative values (red circles) indicate negative correlations. The shade and size of each circle indicate the magnitude of the correlation (with darker shades indicating higher correlations). PCD: Post-cholecystectomy diarrhea; PCND: Post-cholecystectomy non-diarrhea.
The ratio of Firmicutes/Bacteroidetes is an important parameter in evaluating the composition of the intestinal microbiota. The members of Bacteroidetes have been reported to be associated with immunity and metabolic processes[33], and the relative abundance of the phylum Bacteroidetes increased significantly in the PCD group, whereas that of Firmicutes was decreased, which is consistent with the gut microbiota associated with HIV infection[34,35]. In addition, cholecystectomy increases the abundance of Bacteroidetes, a contributor to colorectal cancer[2]. As the abundance of Bacteroidetes was significantly higher in the PCD group than in the PCND group, this increase in Bacteroidetes may be related to diarrhea. Consistent with previous research[2], our investigation showed that the fecal abundance of Bacteroides decreased in PC patients compared to HCs, Furthermore, our investigation indicated that the genus Bacteroides decreased in the PCD group compared with the PCND group, which is consistent with findings for antibiotic-associated diarrhea[36]. Our study also showed that the genus Sutterella was significantly higher in the PCD group than in the PCND group. Research has shown that the abundance of Sutterella in mice with antibiotic-induced diarrhea is significantly higher than in normal mice, indicating that Sutterella may promote diarrhea; after feeding the Chinese medicine Shenqibai to the mice, the structure of the flora was normalized, and the number of Sutterella in the mice decreased significantly[36].

Figure 4 Kyoto Encyclopedia of Genes and Genomes functional categories of microbiota with differential abundance. A: Comparison between the post-cholecystectomy diarrhea-enriched and post-cholecystectomy non-diarrhea-enriched markers regarding the Kyoto Encyclopedia of Genes and Genomes level 2 functional categories; B: Comparison between the post-cholecystectomy diarrhea-enriched and post-cholecystectomy non-diarrhea-enriched markers regarding the Kyoto Encyclopedia of Genes and Genomes level 3 functional categories. PCD: Post-cholecystectomy diarrhea; PCND: Post-cholecystectomy non-diarrhea; LDA: Linear discriminant analysis.
Prevotella is a conditional pathogenic bacterial genus. Research has linked the increased abundance of Prevotella to localized and systemic disease, including periodontitis, bacterial vaginosis, rheumatoid arthritis, metabolic disorders, and lowgrade systemic inflammation[37]. Our study showed a significant increase in the abundance of Prevotella in the PCD group compared to the PCND group. Therefore, Prevotella may also play a role in promoting diarrhea.
Our results suggested a negative correlation between Prevotella and Bifidobacterium, which may be an important cause of diarrhea. We found a higher abundance of Bifidobacterium in the PC group compared with the HC group, which is consistent with previous research[38]. This may reflect the physiological adjustments in the body following loss of the reservoir function of the gallbladder. However, Bifidobacterium was significantly less abundant in the PCD group than in the PCND group. Therefore, lack of Bifidobacterium may have an important effect in PCD. Bifidobacterium is a beneficial genus and is one of the main bacterial groups found on the surface of the normal intestinal mucosa. It is involved in the detachment of bile acid, maintaining the composition of normal intestinal flora, and regulating the immune function of the intestinal mucosa[39-41]. Probiotics including Bifidobacterium have been used in the treatment of dysbacteriosis[42-45]. Bifidobacterium is very important in the balance of bacteria[46-48]. This study showed that Bifidobacterium was significantly reduced in patients with PCD, which provides a theoretical basis for the treatment of PCD using Bifidobacteria probiotics.
Our study also showed that the abundance of Lactococcus was higher in the PCND group than in the PCD group. Lactococcus is a genus in the family Lactobacillaceae and the order Lactobacillales, and it is a beneficial bacteria that can produce lactic acid by fermentation, promote intestinal peristalsis, and prevent constipation and diarrhea[49]. Therefore, we believe that the change in Lactococcus plays a role in the process of PCD.
Using a function analysis based on the KEGG database, we found that lipid metabolism pathways were markedly enriched in the PCND group compared to the PCD group. This may explain why the PCD patients who ate too much fat had frequent diarrhea.
CONCLUSION
In conclusion, we report gut dysbiosis in PCD patients, which is a result of differences in microbial structure, diversity, and abundance when compared to PCND patients. Decreased diversity and abundance of the microbial community in PC patients can cause diarrhea. Our study findings demonstrate the association between PCD and the gut microbiota, especially regarding Prevotella and Bifidobacterium. Thus, this study provides new insights into potential therapeutics that could target the microbiota to attenuate PCD.
ARTICLE HIGHLIGHTS


Research conclusions
These findings demonstrate the association between PCD and the gut microbiota. Gut dysbiosis in PCD patients is a result of differences in microbial structure, diversity, and abundance when compared to PCND patients. Decreased diversity and abundance of the microbial community in PC patients can cause diarrhea.
Research perspectives
This study demonstrated that gut dysbiosis may play a critical role in PCD, especially regarding Prevotella and Bifidobacterium. Thus, this study provides new insights into potential therapeutics that could target the microbiota to attenuate PCD.
 World Journal of Gastroenterology2021年5期
World Journal of Gastroenterology2021年5期
- World Journal of Gastroenterology的其它文章
- Liver injury in the era of COVID-19
- Histopathology and the predominantly progressive, indeterminate and predominately regressive score in hepatitis C virus patients after direct-acting antivirals therapy
- Comparative study of indocyanine green-R15, Child-Pugh score, and model for end-stage liver disease score for prediction of hepatic encephalopathy after transjugular intrahepatic portosystemic shunt
- Vedolizumab in Crohn’s disease with rectal fistulas and presacral abscess: A case report