
Circular RNA AKT3 governs malignant behaviors of esophageal cancer cells by sponging miR-17-5p
2021-02-04 09:46:58HongLiangZangFuJianJiHaiYingJuXiaoFengTian
Hong-Liang Zang, Fu-Jian Ji, Hai-Ying Ju, Xiao-Feng Tian
Abstract
Key Words: Esophageal cancer; Circular RNA AKT3; miR-17-5p; Proliferation; Migration; Invasion
INTRODUCTION
As one of the most malignant tumors, esophageal cancer ranks in the top ten in morbidity and mortality of the tumors worldwide[1]. However, the exact mechanism is still in discussion. Currently, surgery is the predominant approach for treatment of esophageal cancer in combination with some other alternatives[2,3]. Although great progress has been made in the diagnosis and treatment of esophageal cancer, the prognosis has not been improved satisfactorily with a 5-year survival rate of less than 30%[4]. Thus, investigating the underlying mechanism is conducive to deepening the pathogenesis of esophageal cancer and developing targeted therapies.
Circular RNAs (circRNAs) belong to endogenous noncoding RNAs, which could be expressed in eukaryotic cells[5]. Unlike other RNA molecules, circRNAs possess the structure of a closed loop and cannot be degraded by RNase R[6]. Accumulating evidence has demonstrated that circRNAs participate in the modulation of protein expression by sponging microRNAs (miRNAs) or interacting with RNA binding proteins[7,8]. In tumor tissues and cell lines, the aberrant expression of circRNAs is involved in the regulation of malignant phenotypes of tumor cells. Wanget al[9]reported that circLMTK2 is overexpressed in gastric cancer, which could also be a predictor for a poor prognosis in patients with gastric cancer. In colorectal cancer,circFBXW7is significantly downregulated, andcircFBXW1overexpression suppresses proliferative, migratory, and invasive abilities of colorectal cancer cells[10]. Therefore, differential expression of circRNAs exerts the opposite effects on tumor cells. However, only a few studies have concentrated on the effects of circRNAs on esophageal cancer.
Circular RNA AKT3 (circAKT3) is a novel molecule, which is derived from theAKT3gene and identified in gastric cancer with cisplatin resistance[11]. Thein vitroandin vivoassays have revealed that circAKT3 is overexpressed in cisplatin resistantgastric cancer and leads to the increased expression ofPIK3R1to mediate resistance of gastric cancer cells to cisplatin[11]. However, Xiaet al[12]demonstrated that glioblastoma expressed a low level ofcircAKT3, andcircAKT3overexpression suppressed proliferation and promoted sensitivity to radiation of glioblastoma cells. The contradictory findings remind us of the potential role of circAKT3 in esophageal cancer.
MATERIALS AND METHODS
Collection of clinical samples
The clinical tissue samples were acquired from patients with esophageal cancer (n= 82) who underwent surgery at China-Japan Union Hospital of Jilin University. The relative expression ofcircAKT3less than the mean value was classified into the circAKT3 low group (n= 43), and the relative expression ofcircAKT3more than the mean value was classified into the circAKT3 high group (n= 39).
In vivo assay
The nude mice (n= 12) were purchased from Experimental Animal Center of China-Japan Union Hospital of Jilin University, housed in cages under the condition of the 12-h shift of light-dark cycles, and randomly divided into two groups. Negative control short hairpin RNA and short hairpin RNA against circAKT3, purchased from Integrated Biotech Solutions Co. (China), were transfected into TE-1 cells in the presence of puromycin (3 mg/mL; Thermo Fisher Scientific, United States) to establish thecircAKT3knockdown cell lines. The mice injected with the negative control short hairpin RNA-transfected TE-1 cells were designated as the negative control short hairpin RNA group, and mice injected with the short hairpin RNA against circAKT3-transfected TE-1 cells were designated as the short hairpin RNA against circAKT3 group. After feeding for 30 d, the mice were anesthetized with pentobarbital sodium (25 mg/kg; Sigma-Aldrich, United States) intraperitoneally and killed by decapitation. The xenografts were isolated carefully and weighed.
Cell culture
Human esophageal cancer cell lines (KYSE-150, TE-10, TE-1) and human embryonic kidney cell line (HEK-293T) were cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum (Gibco, United States). Human normal esophageal epithelial cell line (HEEC) was cultured in Dulbecco’s Modified Eagle Medium with high glucose containing 10% fetal bovine serum (Gibco, United States). All of the cells were cultured in a CO2incubator chamber (Thermo Fisher Scientific, United States).
Quantitative real time-polymerase chain reaction
TRIzol (Ambion, United States) was utilized to extract the total RNA. SuperScript III (Invitrogen, United States) was used to obtain cDNA. SYBR Premix Ex Taq II (Takara, Japan) was utilized to perform quantitative real time(qRT)-PCR. The primer sequences of circAKT3 and U6 were adopted as previously reported[11]. The primer sequences of miR-17-5p were obtained from recent research[13]. The primer sequences of AKT3, RHOC, STAT3, and GAPDH were obtained from the NCBI online database (https://www.ncbi.nlm.nih.gov/gene/). CircAKT3 (forward, 5’-TCCAAATAAACGCCT TGGTGG-3’; reverse, 5’-CCTCAGAGAACACCCGCTCT-3’); miR-17-5p (forward, 5’-CGGCGGCAAAGTGCTTACAG-3’; reverse, 5’-GTGCAGGGTCCGAGGT-3’); U6 (forward, 5’-CTCGCTTCGGCAGCACA-3’; reverse, 5’-AACGCTTCACGA ATTTGCGT-3’); AKT3 (forward, 5’-CGGCTTTCTCACGGATCACA-3’; reverse, 5’-CGGGACACTTTCCTTCCTCC-3’); RHOC (forward, 5’-AAGTTCCCTTTGCCCG TCTG-3’; reverse, 5’-ACAGTGGCAACTCAAGGGTC-3’); STAT3 (forward, 5’-CCAGGTACCGTGTGTCAAGC-3’; reverse, 5’-CAGACCTGACACCTGTGTTG-3’); GAPDH (forward, 5’-TGAAATGTGCACGCACCAAG-3’; reverse, 5’-GGGAAGCAGCATTCAGGTCT-3’). All of the primers were synthesized by Sangon (China).
Cell transfection
The siRNAs against circAKT3, miR-17-5p mimics, miR-17-5p inhibitor, indicated negative control, and plasmids were synthesized and purchased from Sangon (China). Lipofectamine 3000 (Invitrogen, United States) was used to conduct the transfection. RNase R (Epicentre, United States) was used to digest linear RNAs (but not circRNAs) in line with the manufacturer’s instruction.
Fluorescence analysis
The Hoechst Staining Kit (Beyotime, China) was used to evaluate apoptotic cells. In brief, TE-1 cells were seeded at a density of 2.0 × 105cells per well. After cell transfection with indicated vectors, 4% paraformaldehyde was used to fix the cultured TE-1 cells, followed by using Hoechst 33258 to stain the cells. A fluorescence microscope (Olympus, Japan) was utilized to observe the cells.
Measurement of cell viability
The Cell Counting Kit-8 (CCK-8; Beyotime, China) was utilized to measure cell viability. After transfection, TE-1 cells were seeded at a density of 1 × 104cells per well for the indicated time. CCK-8 solution was added into each well for 2 h, and the Microplate Reader (Bio-Rad, United States) was used to record the value at 450 nm.
Wound healing assay
The wound healing assay was used to measure the migratory ability. After transfection, TE-1 cells were seeded into a 6-well plate for 24 h. The cells were scratched by a 10 mL Finntip and cultured in serum-free medium. Images were captured at 0 h and 24 h and analyzed by ImageJ software.
Measurement of invasive ability
The Transwell assay was conducted to evaluate the invasive ability. TE-1 cells with indicated transfection were cultured in Millicell Standing Cell Culture 24 well (Millipore, United States). After incubation for 24 h, glutaraldehyde solution was used to fix the cells, and crystal violet was used to stain the cells.
Luciferase reporter assay
ENCORI database was used to acquire the predicted binding sites between circAKT3 and miR-17-5p. TargetScanHuman database was utilized to acquire the potential binding sites of miR-17-5p with RHOC or STAT3. The procedures of the luciferase reporter assay were conducted in line with the previous study[11]. In our research, HEK-293T cells were utilized to detect the binding ability by cotransfecting the indicated vectors (Sangon, China) with Lipofectamine 3000 (Invitrogen, United States). The luciferase activity was evaluated by Bio-GloTMLuciferase Assay System (Promega, United States).
Western blot
Western blot was carried out in line with the previous report[14]. In brief, the total protein was lysed by RIPA lysis buffer (Beyotime, China), and the BCA Kit (Thermal Fisher Scientific, United States) was used to detect the precise concentration. The total protein was added into SDS-PAGE gel (Beyotime, China). Primary antibodies [STAT3 rabbit monoclonal antibody (Cell Signaling Technology, United States); RHOC rabbit monoclonal antibody (Cell Signaling Technology, United States); GAPDH mouse monoclonal antibody (Cell Signaling Technology, United States)] were used overnight at 4 °C. Secondary antibodies with horseradish peroxidase (Beyotime, China) were applied for 2 h, and the indicated proteins were monitored by ChemDocTMXRS+ System (Bio-Rad, United States).
Statistical analysis
The data in our research were displayed as mean ± standard deviation and analyzed by GraphPad Prism 8.0 Software (GraphPad, United States). Thet-test, two-way ANOVA or Chi-squared test was used. Statistical significance was set as thePvalue less than 0.05.
RESULTS
CircAKT3 was overexpressed in esophageal cancer
First, we detected the expression ofcircAKT3in the esophageal cancer tissue samples and cell lines. The qRT-PCR results showed that circAKT3 was significantly upregulated in esophageal cancer tissue samples (Figure 1A). The characteristic analysis of the patients with esophageal cancer indicated that the tumor size, lymphatic metastasis, and clinical tumor node metastasis staging were positively related to the upregulation of circAKT3 in the tissue samples (Table 1). We also found that human esophageal cancer cell lines TE-1 cells, TE-10 cells, and KYSE-150 cells expressed a significantly higher level of circAKT3 compared to human normal esophageal epithelial cell line HEEC cells (Figure 1B).
CircAKT3 promoted malignant behaviors of TE-1 cells
Next, siRNAs against circAKT3 were transfected into TE-1 cells, and qRT-PCR analysis revealed a marked efficiency ofcircAKT3knockdown (Figure 1C). As the differential structure between circAKT3 and AKT3 mRNA, circAKT3 was resistant to RNase R,while AKT3 mRNA was sensitive to RNase R (Figure 1C and 1D), which confirmed the circular structure of circAKT3. Moreover, thecircAKT3overexpression plasmids were transfected into TE-1 cells and could significantly increase the expression ofcircAKT3(Figure 1E). CCK-8 assay showed that circAKT3 siRNAs inhibited proliferation of TE-1 cells, whilecircAKT3overexpression plasmids facilitated proliferation of TE-1 cells (Figure 1F). We also found thatcircAKT3knockdown suppressed migration and invasion of TE-1 cells, whilecircAKT3overexpression exerted the opposite role by conducting the wound healing assay and Transwell assay, respectively (Figure 1G and 1H). We also measured the effect of circAKT3 on apoptosis. The fluorescence analysis indicated that circAKT3 siRNAs increased the number of condensed nuclei in TE-1 cells; however,circAKT3overexpression had no effect on the apoptotic rate of TE-1 cells (Figure 1I). These findings suggested that circAKT3 facilitated proliferative, migratory, and invasive capacities of esophageal cancer cells.

Table 1 Characteristics of patients with esophageal cancer
miR-17-5p acted as the downstream molecule of circAKT3 to antagonize the effects of circAKT3 on TE-1 cells
Accumulating evidence has reported that circRNAs regulate cell functions by sponging miRNAs[7]. The ENCORI database (http://starbase.sysu.edu.cn/index.php) was utilized to predict the potential target of circAKT3, and we identified miR-17-5p as the target of circAKT3. We carried out a luciferase reporter assay and found that TE-1 cells with wild-type circAKT3 and miR-17-5p mimics cotransfection presented a remarkably decreased luciferase activity (Figure 2A). Next, we detected the expression ofmiR-17-5pand found a lower level of miR-17-5p in esophageal cancer tissue samples (Figure 2B). Moreover,miR-17-5pexpression was inversely associated with the expression ofcircAKT3in clinical tissue samples (Figure 2C). Meanwhile, esophageal cancer cell lines showed decreased expression ofmiR-17-5p(Figure 2D). These results indicated that miR-17-5p was inhibited by circAKT3. We then explored the role of miR-17-5p functioning as a downstream molecule of circAKT3 in TE-1 cells. CCK-8 assay showed that cell viability of TE-1 cells with cotransfection ofcircAKT3overexpression plasmids and miR-17-5p mimics was decreased compared to that withcircAKT3overexpression plasmids transfection (Figure 2E). Moreover, miR-17-5p mimics suppressed migratory and invasive abilities ofcircAKT3overexpression plasmids-transfected TE-1 cells by the wound healing assay and Transwell assay, respectively (Figure 2F and 2G). In parallel, miR-17-5p mimics increased the apoptotic rate ofcircAKT3overexpression plasmids-transfected TE-1 cells (Figure 2H).
RHOC and STAT3 were modulated by the circAKT3/miR-17-5p axis


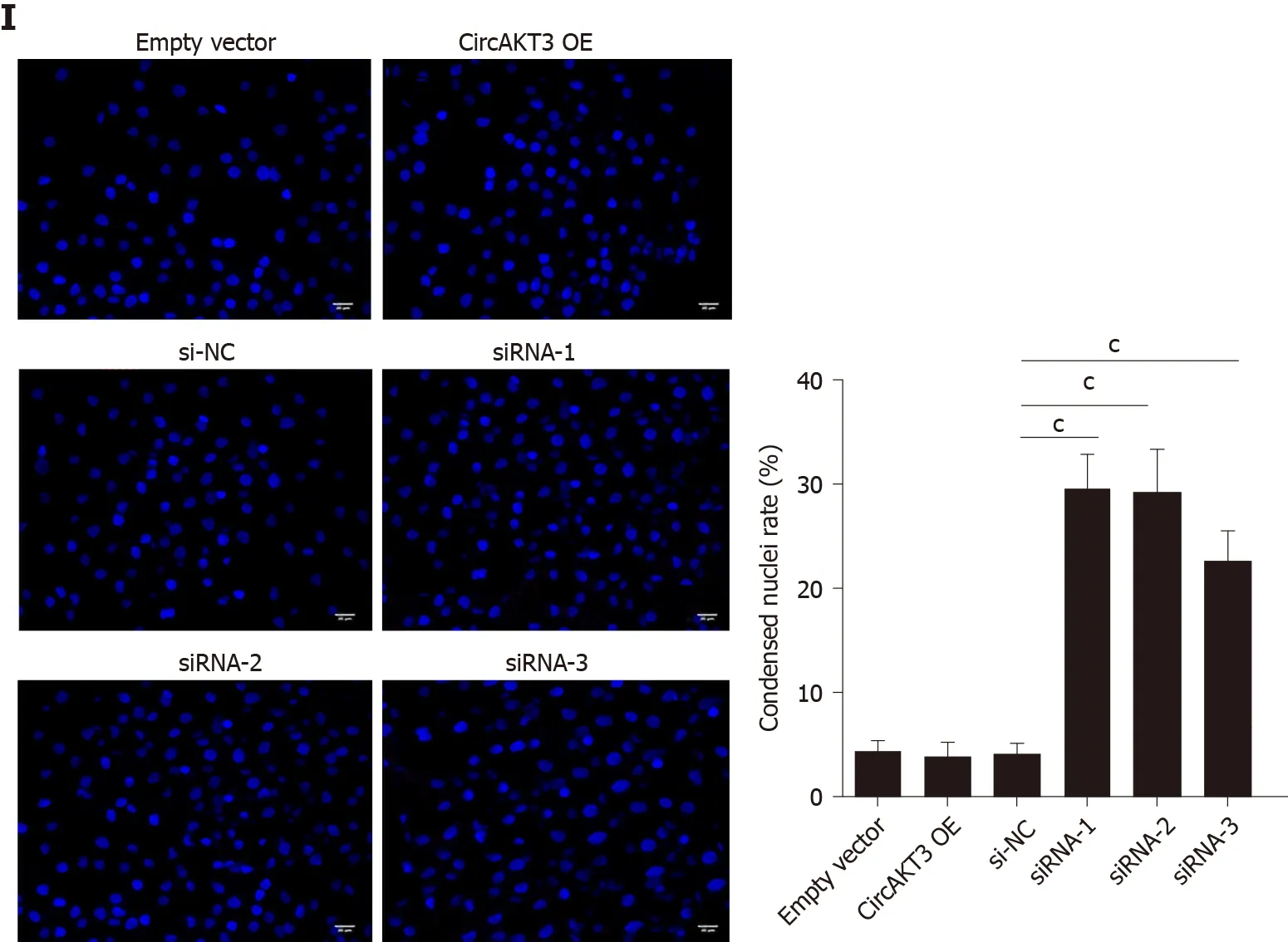
Figure 1 Circular RNA AKT3 promoted malignant phenotypes of TE-1 cells. A: The expression of circular RNA AKT3 (circAKT3) in tissue samples; B: The expression of circAKT3 human normal esophageal epithelial cell line (HEEC) and human esophageal cancer cell lines; C and D: TE-1 cells were transfected with control oligo and circAKT3 siRNAs in the presence or absence of RNase R to detect the expression of circAKT3 and AKT3; E: The expression of circAKT3 in TE-1 cells with circAKT3 overexpression (OE); F: Cell viability of TE-1 cells as detected by the Cell Counting Kit-8 assay was performed to detect cell viability of TE-1 cells with indicated transfection; G: Migratory capacity of TE-1 cells with circAKT3 OE and knockdown; H: Invasive ability of TE-1 cells with circAKT3 OE and knockdown; I: Apoptosis of TE-1 cells with circAKT3 OE and knockdown by Hoechst staining. aP < 0.05; bP < 0.01; cP < 0.001. NC: Negative control; OD: Optical density.
We then identified RHOC and STAT3 as the potential targets of miR-17-5p by using TargetScanHuman database (http://www.targetscan.org/vert_72/). The luciferase reporter assay showed that TE-1 cells with cotransfection of wild-type RHOC and miR-17-5p mimics presented a lower luciferase activity (Figure 3A). In addition, the decreased luciferase activity was also shown in TE-1 cells with cotransfection of wildtype STAT3 and miR-17-5p mimics (Figure 3B). Next, we detected the mRNA level of RHOC and STAT3 in clinical samples and found that both RHOC and STAT3 were upregulated in esophageal cancer tissue samples (Figure 3C and 3D). Moreover, the expression ofRHOCandSTAT3was increased in TE-1 cells, TE-10 cells, and KYSE-150 cells (Figure 3E and 3F). Western blot analysis showed that circAKT3 promoted the expression of RHOC and STAT3 in TE-1 cells, and miR-17-5p led to the reduced expression of RHOC and STAT3, which was reversed by circAKT3 overexpression plasmids cotransfection (Figure 3G). Conversely, circAKT3 siRNA inhibited the expression of RHOC and STAT3 in TE-1 cells, and a miR-17-5p inhibitor promoted the expression of RHOC and STAT3, which was reverted by circAKT3 siRNA cotransfection (Figure 3H). In consequence, circAKT3 facilitated the expression of RHOC and STAT3 by sponging miR-17-5p in esophageal cancer cells.
CircAKT3 knockdown suppressed growth of esophageal cancer in vivo
CircAKT3 shRNA was transfected into TE-1 cells to establish the stablecircAKT3knockdown cell line (Figure 4A). Thein vivoassay showed that the size and weight of the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells were smaller and lighter (Figure 4B and 4C). The qRT-PCR analysis indicated that the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells expressed a lower level of circAKT3, RHOC, and STAT3 and a higher level of miR-17-5p (Figure 4D-G). In parallel, the protein levels of RHOC and STAT3 were decreased in the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells (Figure 4H).
DISCUSSION


Figure 2 miR-17-5p acted as the downstream molecule of circular RNA AKT3 to antagonize the effects of circular RNA AKT3 on TE-1 cells. A: Schematic of the miR-17-5p binding sites in wild-type (WT) circular RNA AKT3 (circAKT3) and the mutant (Mut) circAKT3 (up). Luciferase reporter assay was performed in HEK-293T cells to determine the binding ability between miR-17-5p and circAKT3 (down); B: The expression of miR-17-5p in tissue samples; C: CircAKT3 was negatively correlated with miR-17-5p in esophageal cancer tissue samples; D: miR-17-5p expression in indicated cell lines; E-H: Cell viability (E), migratory ability (F), invasive ability (G), and apoptosis (H) of circAKT3 overexpression (OE)-transfected TE-1 cells in the presence or absence of miR-17-5p mimics. a P < 0.05; bP < 0.01; cP < 0.001. HEEC: Human normal esophageal epithelial cell line; NC: Negative control; OD: Optical density.
Increased proliferative, migratory, and invasive abilities are hallmarks of tumor cells[15]. Thus, exploring the exact mechanism of malignant phenotypes of tumor cells contributes to clarifying the pathogenesis of tumorigenesis. Herein, we reported the protumor role of circAKT3 in esophageal cancer cells by measuring the proliferative, migratory, and invasive abilities. In addition, miR-17-5p acted as a sponge molecule to inhibit the effects of circAKT3 on esophageal cancer cells. Furthermore, two protumor molecules, RHOC and STAT3, were the direct downstream targets of miR-17-5p, and circAKT3 upregulated RHOC and STAT3 by inhibiting miR-17-5p in esophageal cancer cells. Finally, we foundcircAKT3knockdown inhibited growth of esophageal cancerin vivo.
CircRNAs function among the complex molecular network and are implicated in the expression of functional proteins[5,7]. Mechanistically, the inhibitory effect of circRNAs on miRNAs is one of the predominant mechanisms involving regulation of proteins[7]. Increasing evidence has focused on the effects of circRNAs on the modulation of malignant phenotypes of tumor cells; however, only a few studies explore the relationship between circRNAs and esophageal cancer. A recent study reported that circZNF292 is upregulated in the esophageal cancer cell line Eca-109, and inhibition of circZNF292 enhanced the expression ofmiR-206to limit the proliferative, migratory, and invasive ability of Eca-109 cells[16]. In addition, Shiet al[17]demonstrated that overexpressed circPRKCI promoted the expression ofAKT3by inhibiting miR-3680-3p to facilitate proliferation and migration of esophageal cancer cells. In addition, circSMAD7[18]and circFOXO3[19]could sponge different miRNAs to suppress malignant phenotypes of esophageal cancer cells. Therefore, it can be inferred that circRNAs exert the opposite roles in development and progression of esophageal cancer.
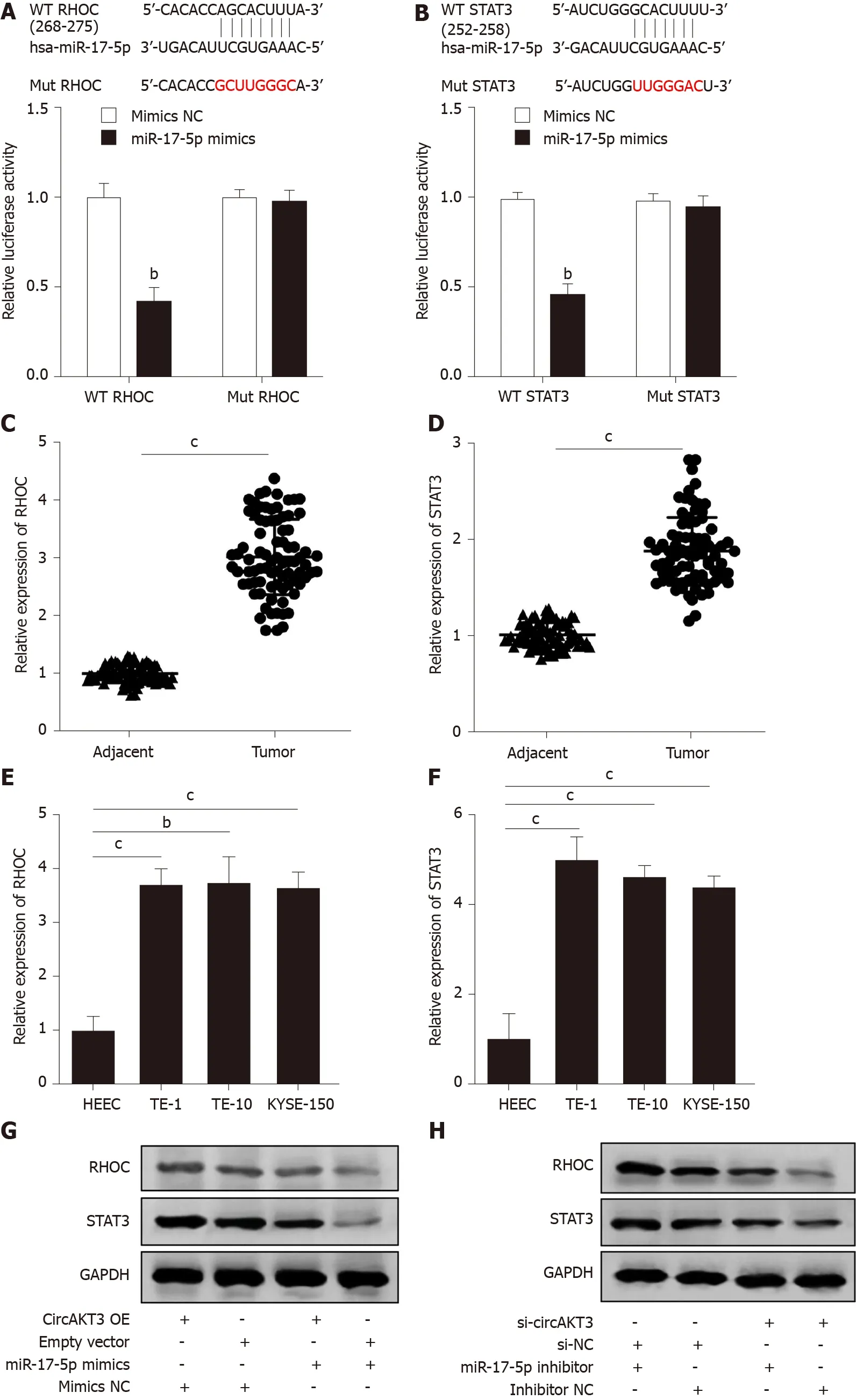
Figure 3 RHOC and STAT3 were modulated by the circular RNA AKT3/miR-17-5p axis. A: Schematic of the miR-17-5p binding sites in wild-type (WT) RHOC and the mutant (Mut) RHOC (up). Luciferase reporter assay was performed in HEK-293T cells to determine the binding ability between miR-17-5p and RHOC (down); B: Schematic of the miR-17-5p binding site in WT STAT3 and the Mut STAT3 (up). Luciferase reporter assay was performed in HEK-293T cells to determine the binding ability between miR-17-5p and STAT3 (down); C and D: The expression of RHOC (C) and STAT3 (D) in tissue samples; E and F: The expression of RHOC (E) and STAT3 (F) in indicated cell lines; G and H: Protein level of RHOC and STAT3 in TE-1 cells with indicated transfection. bP < 0.01; cP < 0.001. circAKT3: Circular RNA AKT3; HEEC: Human normal esophageal epithelial cell line; NC: Negative control; OE: Overexpression.
Our research identified circAKT3 as a motivator in development and progression of esophageal cancer cells. The gene symbol of circAKT3 isAKT3, which is a key member of the PI3K/AKT signaling pathway[11,20]. Previously, Huanget al[11]reported thatcircAKT3overexpression in gastric cancer participated in cisplatin resistance by restraining miR-198. However, the contradiction about the effects of circAKT3 on tumors is that circAKT3 could suppress development of glioblastoma[12]and progression of clear cell renal cell carcinoma[20]. These findings, combined with our results, indicate a tumor specificity of circAKT3. Moreover, we identified miR-17-5p as a downstream molecule of circAKT3 to suppress the effects of circAKT3 on esophageal cancer cells by evaluating proliferation, migration, and invasion of TE-1 cells and found that esophageal cancer tissue samples and cell lines showed a significantly lower level of miR-17-5p. It has been reported that the expression ofmiR-17-5pdecreased esophageal cancer stem-like cells and mediated radiation resistance[13], which further reinforced our findings.
Additionally, we screened two protumor molecules RHOC and STAT3 as the direct downstream targets of miR-17-5p. RHOC is one of the Ras homolog family members and functions as a protumor molecule in multiple tumors[21,22]. Clinical data shows that RHOC is positively expressed in esophageal cancer tissue samples and could be regarded as a predictor for patients with a poor prognosis of esophageal cancer[23,24]. Thein vitroandin vivoexperiments manifest that RHOC enhanced the proliferative and invasive capacity of esophageal cancer cells[25]. Similar to RHOC, STAT3 is a pivotal molecule that accelerates development and progression of malignant tumors[26,27]. In esophageal cancer cells, activation of STAT3 dramatically reduced apoptotic rate and contributed to increased proliferative ability[28]. Moreover, STAT3 promoted the expression of programmed death ligand 1 to mediate immune evasion of esophageal cancer cells[29].
CONCLUSION
Taken together, our research discovered that circAKT3 enhanced proliferative, migratory, and invasive capacities of esophageal cancer cells by sponging miR-17-5p to exert protumor effects, and downregulation of circAKT3 inhibited xenograft tumor growth of esophageal cancerin vivo, providing a potential target for better understanding the underlying mechanism of esophageal cancer.

Figure 4 Circular RNA AKT3 knockdown suppressed growth of esophageal cancer in vivo. A: The expression of circular RNA AKT3 (circAKT3) in TE-1 cells with circAKT3 short hairpin (sh)RNA transfection; B and C: The size (B) and weight (C) of the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells; D-G: The expression of circAKT3 (D), miR-17-5p (E), RHOC (F) and STAT3 (G) in the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells by quantitative real time-polymerase chain reaction; H: The protein levels of RHOC and STAT3 in the xenograft tumor injected with circAKT3 shRNA-transfected TE-1 cells. cP < 0.001. sh-NC: Negative control shRNA; sh-circAKT3: Circular RNA AKT3 shRNA.
ARTICLE HIGHLIGHTS


Research results
In vitroassays results revealed that proliferative, migratory, and invasive capacities of esophageal cancer cells were significantly enhanced bycircAKT3overexpression. miR-17-5p was screened as the target of circAKT3, and miR-17-5p antagonized the effects of circAKT3 on esophageal cancer cells. Moreover, we identified RHOC and STAT3 as the direct target molecules of miR-17-5p, and circAKT3 facilitated expression ofRHOCandSTAT3by inhibiting miR-17-5p.In vivoassays showedcircAKT3knockdown inhibited growth of esophageal cancer.
Research conclusions
CircAKT3 enhanced proliferative, migratory, and invasive capacities of esophageal cancer cells by sponging miR-17-5p to exert protumor effects, and downregulation of circAKT3 inhibited xenograft tumor growth of esophageal cancerin vivo, providing a potential target for better understanding the underlying mechanism of esophageal cancer.
Research perspectives
The role of circAKT3 in other tumors may be uncovered in the future, and its application in anticancer therapy will be promoted.
 World Journal of Gastroenterology2021年3期
World Journal of Gastroenterology2021年3期
- World Journal of Gastroenterology的其它文章
- Peritoneal dissemination of pancreatic cancer caused by endoscopic ultrasound-guided fine needle aspiration: A case report and literature review
- Comparative study on artificial intelligence systems for detecting early esophageal squamous cell carcinoma between narrow-band and white-light imaging
- Trends in the management of anorectal melanoma: A multi-institutional retrospective study and review of the world literature
- Serum vitamin D and vitamin-D-binding protein levels in children with chronic hepatitis B
- Screening colonoscopy: The present and the future