
基于煤气化废水处理流程萃取剂选择的量子力学计算
2021-02-02 07:48王绍光徐环斐田文德
青岛科技大学学报(自然科学版) 2021年1期
孔 毅,崔 哲,王绍光,徐环斐,田文德
(青岛科技大学 化工学院,山东 青岛266042)
煤炭气化行业仍然是人们获取能源不可缺少的途径[1]。当前国内外应用最多的煤气化技术为LURGI[2]加压煤气化工艺,具有原料适应性较好、气化强度大、耗氧率低、效率高等优点。然而,煤炭转化成各种产品的同时也产生大量的废水[3]。由于煤气化废水水质成分复杂,污染物浓度高,毒性大[4]。因此,高效处理煤气化废水显得尤为重要。2018年ZHU 等[5]综述了煤气化废水的各种预处理、生物处理和先进处理方法,并对其优缺点进行了比较。2019年LIU 等[6]提出了混凝臭氧法处理煤气化废水的方法。由于煤气化废水的主要污染物为酚类物质,因此处理含酚废水是处理煤气化废水的关键。2017年FENG 等[7]对各种酚类萃取剂的选择进行了研究和比较,选出了最佳的萃取剂,但并没有对萃取机理进行充分解释。2019年GAI等[8]利用乙醇胺(C2H7NO)从煤焦油中分离酚类化合物,并利用量子化学方法研究其萃取机理。
萃取剂的选择仍然依赖于实验方法,这是一个费时费力的过程。因此,有必要寻找一种方便、省时的方法来筛选萃取剂。幸运的是,利用量子力学可以完全理解和预测所研究系统的性质。该方法对研究材料的微观结构与宏观性能之间的关系也有一定的 应 用 前 景 。HARTREE 等[9]在20 世 纪30 年 代提出了独立粒子模型,并将其应用于原子结构的模拟。在之后发展起来的密度泛函理论(Density Functional Theory,DFT)是一种研究多电子系统电子结构的方法。2018 年ZHANG 等[10]使用DFT定量研究了静电相互作用和π-π相互作用,由于忽略了环氧基团的影响,模拟结果与实验结果存在偏差。ANJU 等[11]将DFT 和实验结果相结合,共同解释了研究体系与水的氢键作用。
过程模拟是研究和开发新型工业过程的有效方法。XIAO 等[12]提出了一种利用碱性有机废水促进煤气化的工艺,并利用Aspen Plus进行了一体化的工艺模拟,通过实验数据对模型进行了验证。WANG 等[13]提出了“氨蒸馏-浓缩-超临界水气化-氧化”联合工艺处理高浓度含酚煤气化废水,并通过仿真模拟与实验结果对其经济价值进行了分析。
本工作选用萃取效果较优与溶解度较低的萃取剂甲基丙基酮(MPK)和甲基异丁基酮(MIBK),通过Aspen Plus软件模拟结果发现MPK有良好的萃取效果,出水总酚浓度为66.45 mg·L-1,实现了出水总酚浓度低于300 mg·L-1的净化目标。然后使用量子化学的方法研究萃取精馏中萃取剂的选择问题,以微观分子间相互作用解释宏观特定萃取剂分离效果。依托量子化学高效、低成本的优势,应用于传统的萃取剂选择问题,试图开发用于大量复杂的萃取剂筛选的有效方法。以降低萃取精馏开发过程的成本,减少大量繁琐的萃取剂初步筛选时间,对于萃取精馏过程萃取剂的选择开发具有重要的理论和现实意义。
1 煤气化废水处理流程模拟
1.1 流程概述
近十年来,Aspen Plus是集流程模拟和废水处理为一身的建模和仿真工具[14]。针对含酚废水净化处理工艺过程的研究调查,本设计对废水净化工艺流程分隔为两个工段:煤气化反应工段和萃取脱酚工段。为了进行煤气化废水的生成过程仿真,从某煤化工装置中获得了温度、压力等运行参数。废水净化工艺的简化流程图如图1所示。
1.1.1 煤气化反应工段
用Aspen Plus软件对煤气化废水的生成过程进行了仿真。在Aspen物理性质数据库中,煤、炭、灰分和焦油非常规组分需要首先被考虑。无论是在化学性质还是在相平衡,Aspen Plus都不能直接处理由复杂分子组成的煤。目前,大多数研究都选择了Peng Robinson-Boston Mathias(PR-BM)属性法进行煤气化模拟[15]。在煤气化过程中,利用产率反应器模块[16]对干燥单元和热解单元进行了模拟。干燥单元的温度和压力分别设置为350 ℃和3.55 MPa。通过组分分离器1 去除大部分水分得到干煤。热解装置温度和压力分别设定为630 ℃和3 MPa后,继续通过组分分离器2对热解气体和焦炭进行分离。然后将焦炭引入化学计量反应器中得到纯碳。热解气进一步冷凝,通过组分分离器3得到焦油和煤气化废水。
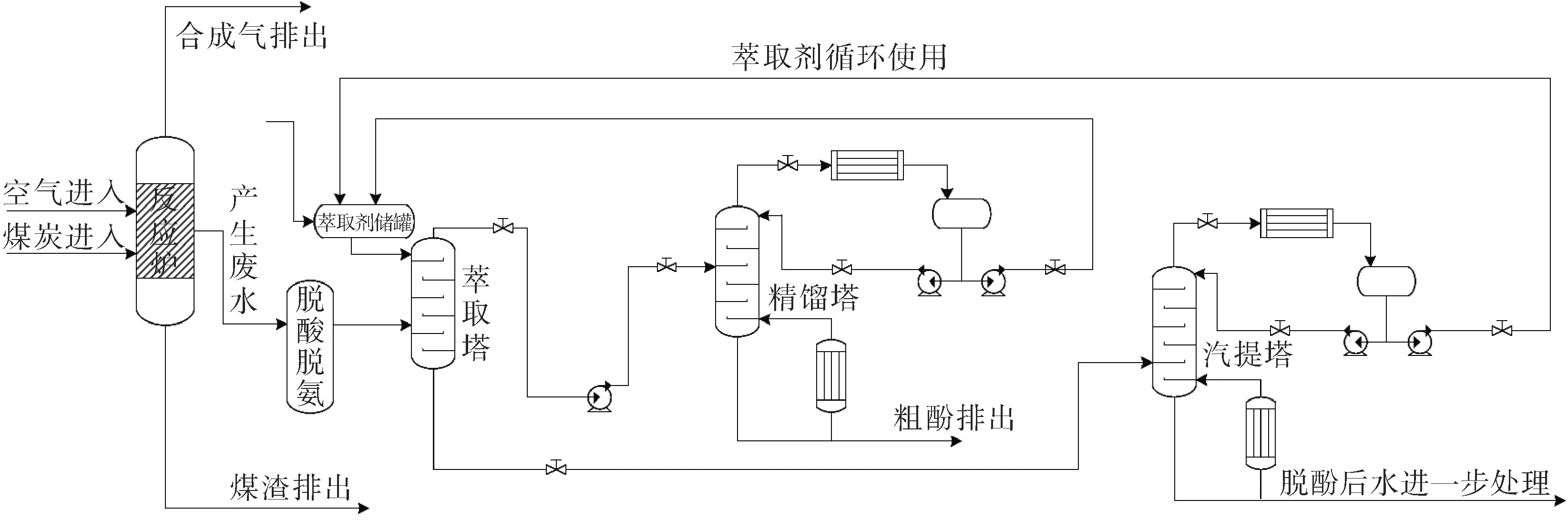
图1 废水净化工艺全流程图Fig.1 General process of wastewater purification process
1.1.2 萃取脱酚工段
煤气化废水依次通过萃取塔,精馏塔和汽堤塔模型进行处理,其中精馏过程采用严格计算模块,并采用NRTL物性方法。萃取剂从塔顶进入与废水在萃取塔中逆流接触,从萃取塔顶部流出的萃取剂及苯酚进入精馏塔,实现萃取剂的循环,萃取塔底部经萃取剂处理后的废水进入汽堤塔得到净化水。其中萃取塔、精馏塔和汽提塔的操作参数如表1所示。
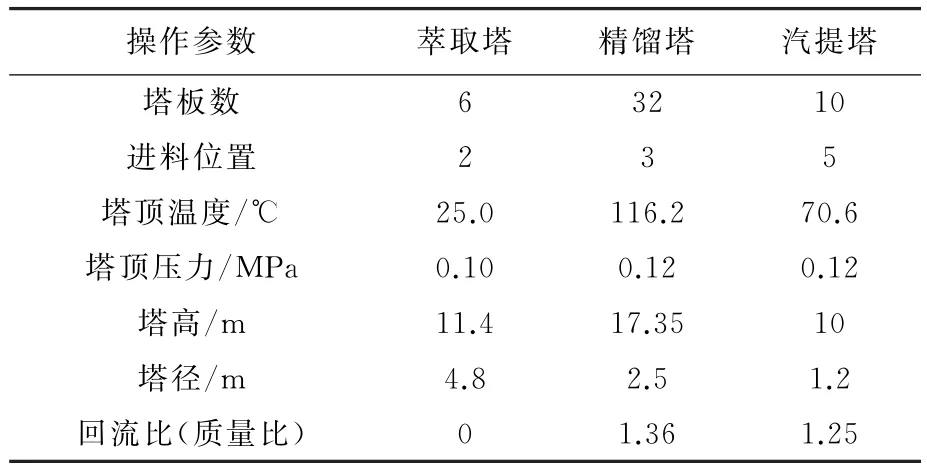
表1 三塔的常用操作参数Table 1 Common operation parameter of three columns
1.2 模拟结果
废水净化工艺的全流程模拟结果如表2~表5所示,萃余相组成如表6所示。
从表2~表5模拟的结果可知,最终分离后的出水总酚浓度为66.45 mg·L-1,符合国家环境保护部出水总酚浓度小于300 mg·L-1的要求,从而可以进行进一步生物处理。
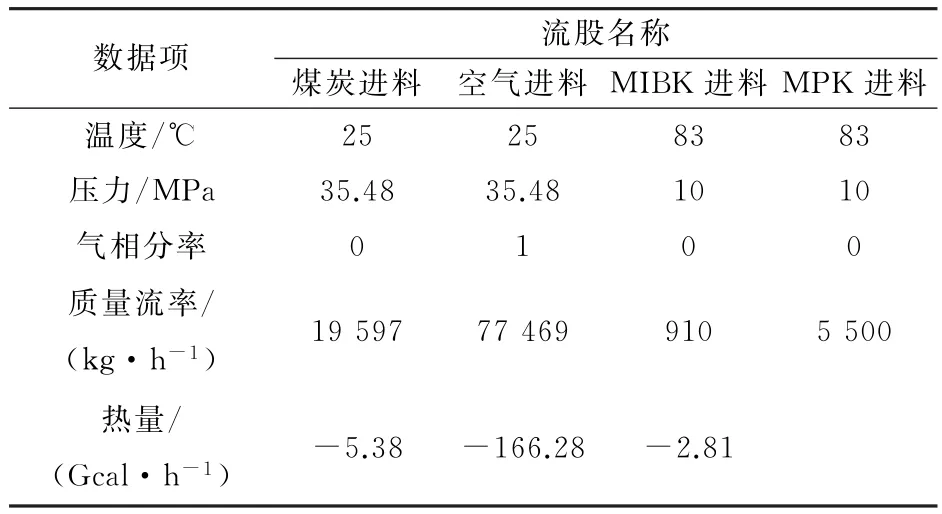
表2 废水处理流程进口物料模拟结果Table 2 Simulation results of input flow in wastewater treatment process
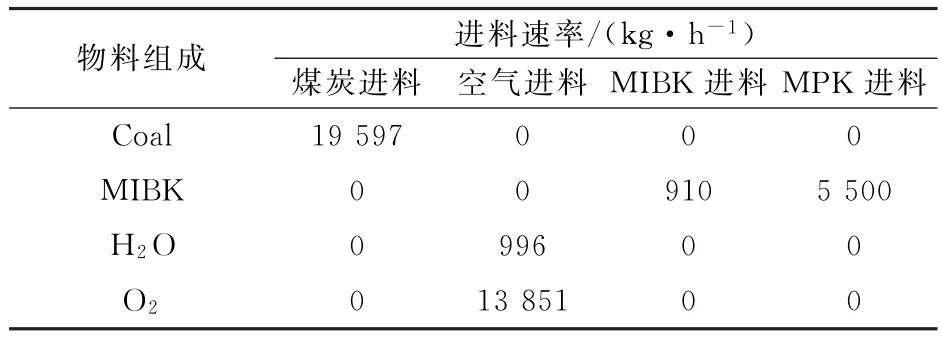
表3 废水处理流程进口物料成分Table 3 Composition of materials at the inlet of wastewater treatment process
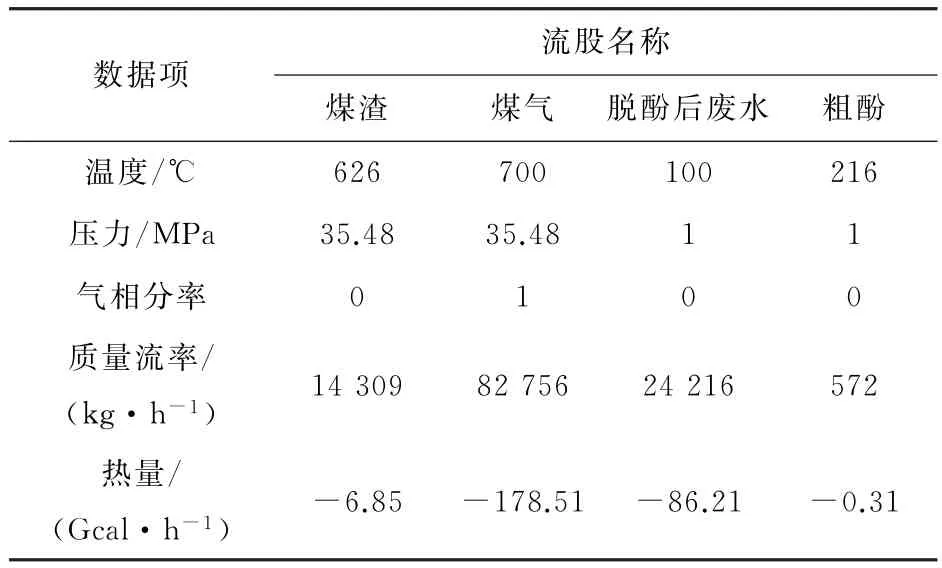
表4 废水处理流程出口物料模拟结果Table 4 Simulation results of output flow in wastewater treatment process
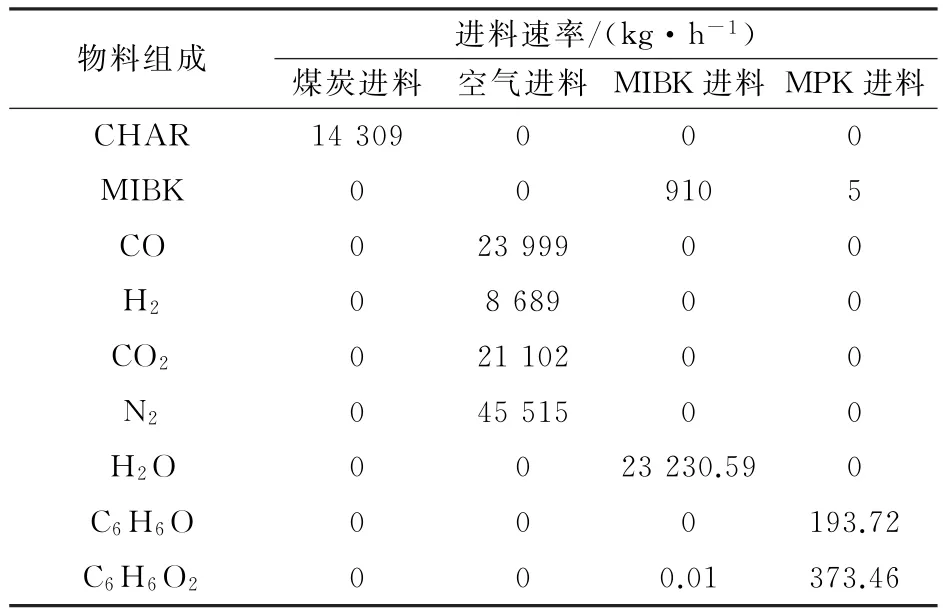
表5 废水处理流程出口物料成分Table 5 Composition of materials at the outlet of wastwater treatment process
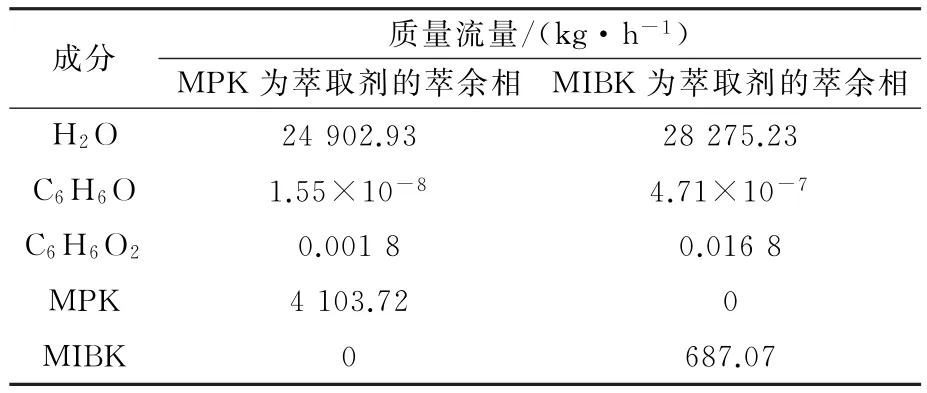
表6 萃余相组成表Table 6 Treatment results of two extractants
从表6可以看出,两种萃取剂对酚类废水均有较好的萃取能力,但MPK 的处理效果优于MIPK的处理效果。
2 量子力学计算相互作用能
2.1 研究方法
实现萃取分离过程的微观本质是研究萃取剂分子和待分离组分分子间的相互作用差异。由于萃取过程为物理过程,没有键的断裂或生成,所以只需要研究体系中分子间的相互作用,即范德华作用、π-π堆积作用、氢键、二氢键、卤键等弱相互作用。
为了定量分析这一相互作用,引入了添加BSSE校正的相互作用能EBSSE这一概念。以双分子A、B为例,其相互作用能如公式1、2和3所示:
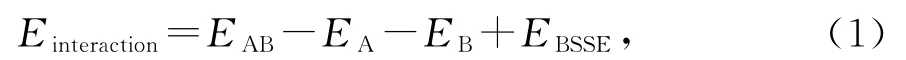
其中:
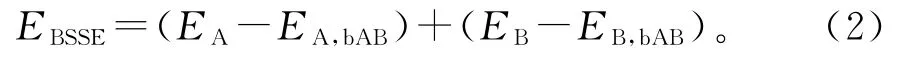
化简后得到:
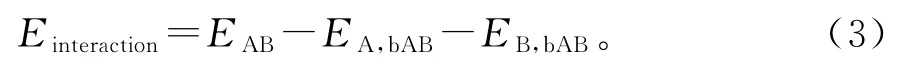
式(1)~(3)中,EAB:A、B 基函数下AB 复合物的能量;EA,bAB:A、B基函数下A 的能量;EB,bAB:A、B基函数下B 的能量;EA:A 基函数下A 的能量;EB:B基函数下B的能量。
2.2 计算过程
本章的相关计算主要使用Gaussian09 D01[17]软件包,同时还使用了Molclus[18]和MOPAC[19]程序。首先,利用Molclus程序寻找体系的全部构象。调用MOPAC程序并使用PM6-DH+进行预优化,继续调用Gaussian 并使用B3LYP 交换泛函结合6-311G(d,p)基组进行再优化。为了消除色散效应,在路径选项栏(route section)中添加Empirical dispersion=GD3BJ关键词,所有计算均在不受对称约束的情况下进行优化。对优化后的结构进行B3LYP/6-311G(d,p)级别下的频率分析,验证能量最小值,并提供零点修正能。在B3LYP/6-311++G(d,p)级别下,添加关键词counterpoise=2 计算BSSE校正下的相互作用能。
2.3 计算结果
采用甲基丙基酮(MPK)、甲基异丁基酮(MIBK)作为萃取剂萃取废水中的苯酚。MIBK/苯酚和MPK/苯酚(物质的量的比1∶1)的稳定构象如图2所示。体系能量与相互作用能如表7所示。
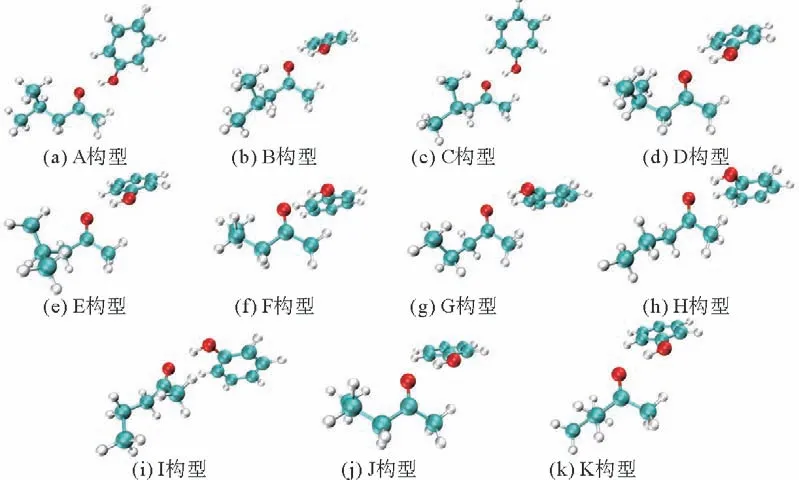
图2 体系在B3LYP/6-311G(d,p)等级下的优化结构构型图Fig.2 Optimized configurations for complex calculated at the B3LYP/6-311G(d,p)level
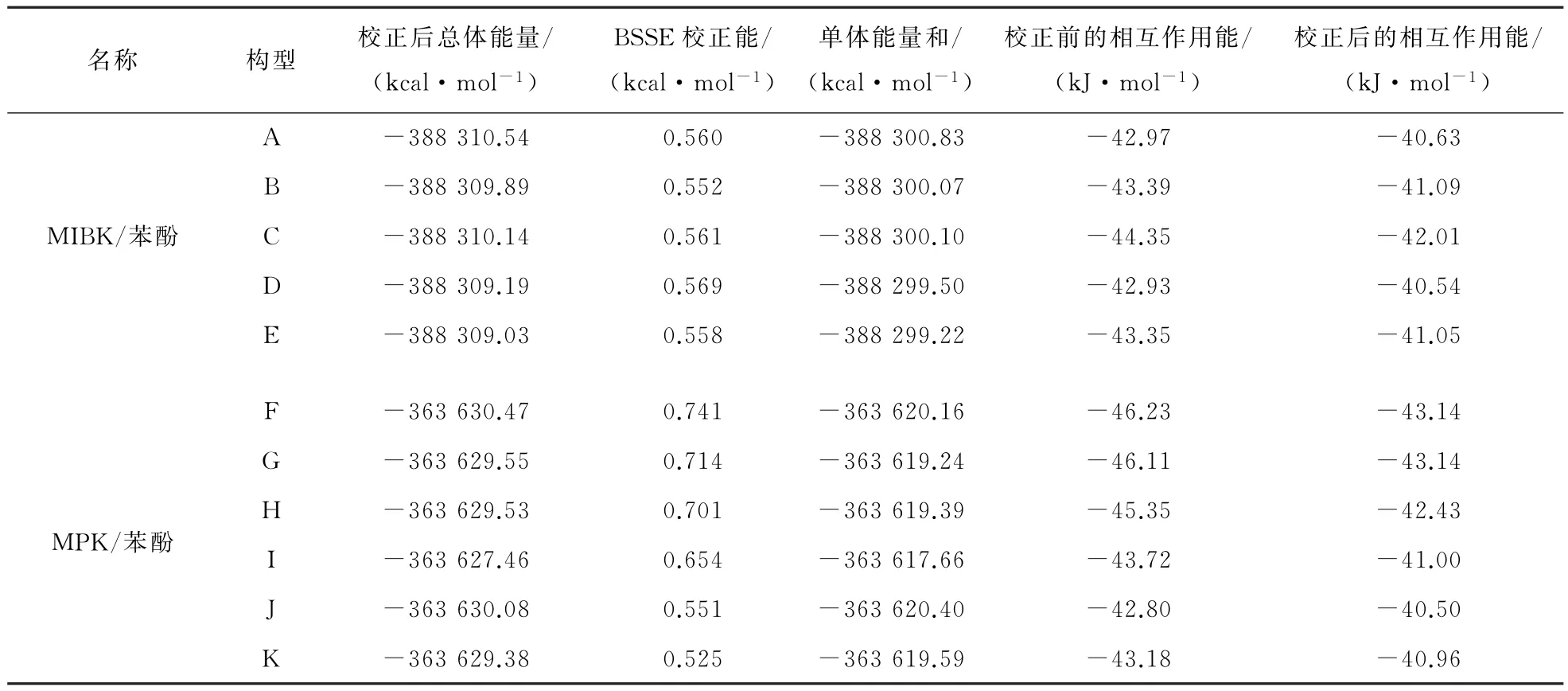
表7 A~F结构构型的能量与相互作用能Table 7 Energy and interaction energy of A—F configurations
相同体系之间的能量差异主要是由不同构象的MIBK(MPK)所产生的。从表7可以看出,MIBK/苯酚的A~E 5种构型中,构型A 的能量最低,结构最稳定,构型C 的分子间相互作用能最大,萃取效果最好;MPK/苯酚的F~K 6种构型中,构型F 不仅能量最低,结构最稳定,而且分子间相互作用能最大,萃取效果最好。最后采用A、C、F、J 4种构型进行对比分析,探究其微观原理。
2.4 RDG 分 析
约化密度梯度分析(reduced density gradient analysis,RDG)是基于电子密度和电子密度梯度来检测实空间函数中非共价的相互作用[20],可以形象地体现出体系中范德华力、氢键、位阻排斥等弱相互作用的区域。约化密度梯度如公式4所示:
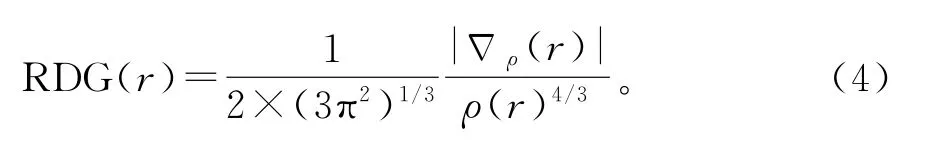
当前工作通过Multiwfn[21]和VMD[22]完成的。
以MIBK/苯酚A、C 两种构型为代表的RDG示意图如图3 所示。在散点图中,纵坐标为RDG函数,横坐标为sign(λ2)ρ(r),λ2为电子密度的Hessian矩阵的第二特征值。ρ(r)可以反映出作用的强度,sign(λ2)可以反映出作用的类型。A 区域(sign(λ2)ρ(r)<0),表示吸引作用较强的区域,比如氢键、卤键等。B 区域(sign(λ2)ρ(r)≈0),表示相互作用很弱,符合范德华作用的特征。C 区域(sign(λ2)ρ(r)>0),表示排斥作用较强的区域。
根据AIM 理论,认为氢键的键临界点处的电子密度为-0.002~-0.037(a.u.)。由图3 可知,3D图中的A 对应散点图靠左的spike,体现为较强的羰基氧与羟基的O—HO 氢键;B对应散点图中间的spike,体现为较弱的相互作用;苯环中间的C区域对应散点图最右边的spike,体现为较强的位阻效应。
在图3的2D 图中,MIBK/苯酚的A 构型在横坐标从-0.015到-0.01的区域内存在一个spike,体现为MIBK 分子内弱氢键作用,可能为A 构型比C构型能量稍低的原因之一;C 构型中间出现两个spike,体现为MIBK 与苯酚的分子间范德华作用,可能为A 构象比C 构象相互作用能稍高的原因之一。MPK/苯酚F构型比J构型的O—HO 氢键的sign(λ2)ρ(r)值大,同时在F构型的3D 图中分子间的B区域比J构型多,体现为分子间弱氢键作用或者范德华作用,为F构型比J构型相互作用能低的原因之一。
2.5 AIM 分 析
分子中的原子理论(quantum theory of atoms in molecules,QTAIM)[23]是BADERΔ提 出 的,基 于电子密度(ρ(r))值和拉普拉斯密度(2ρ(r))来准确理解原子间成键特性。AIM 理论定义了临界点(critical point,CP)的概念,相当于某个函数的梯度的模为0的点。AIM 分析中通常讨论的是电子密度这个函数的临界点。临界点在稳定的电荷分布下可分为(3,-3)、(3,-1)、(3,+1)、(3,+3)4种,其中(3,-3)对应函数的局部极大点,通常出现在离原子核附近,称为核临界点(NCP);(3,-1)对应函数的二阶鞍点,通常出现在有相互作用的两个原子之间,称为键临界点(BCP);(3,+1)对应函数一阶鞍点,通常出现在环体系平面中,称为环临界点(RCP);(3,+3)对应函数的局部极小点,通常出现在笼状体系中,称为笼体系的临界点(CCP)。
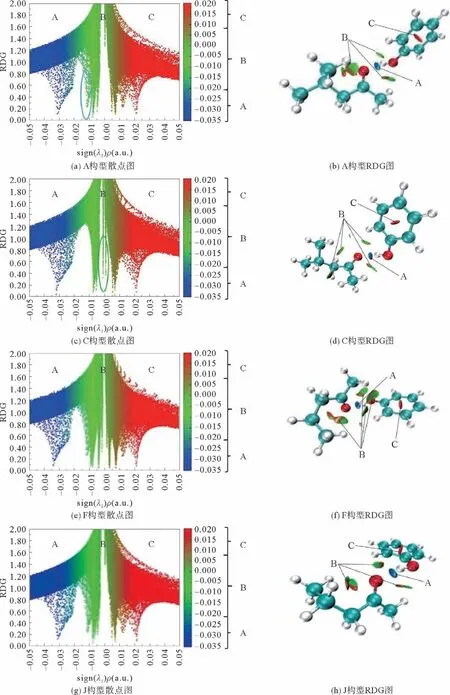
图3 MIBK/苯酚(A、C)和MPK/苯酚(F、J)的RDG图形Fig.3 RDG diagram of MIBK/phenol(A,C)and MPK/phenol(F,J)
基于AIM 理论,提出氢键的存在标准:BCP处的电子密度(ρΔBCP)在0.002~0.035(a.u.)范围内,拉普拉斯密度(2ρBCP)在0.024~0.139范围内并且原子之间距离小于0.35 nm。
根据AIM 理论,使用Multiwfn软件,计算体系中氢键的强弱,列于表8中,并在图4中显示体系中临界点与键路径的位置。
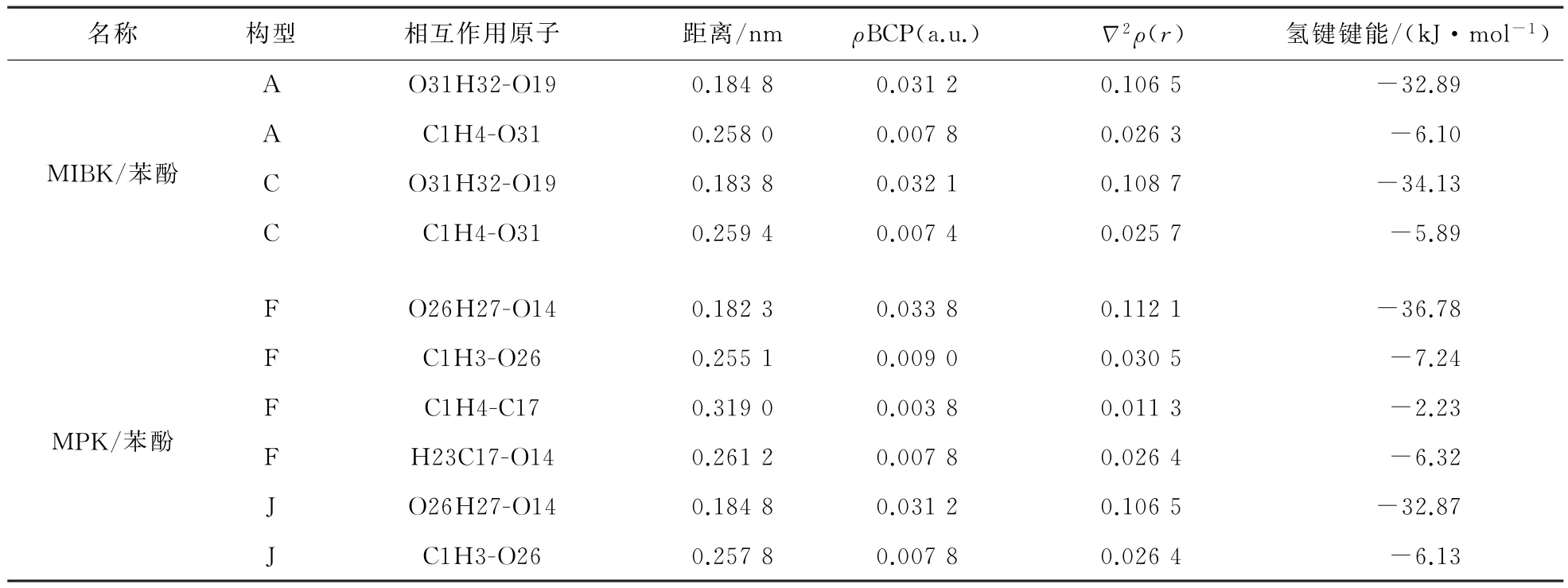
表8 MIBK/苯酚(A、C)和MPK/苯酚(J、K)中氢键的AIM 参数Table 8 AIM parameters of hydrogen bond in MIBK/phenol(A,C)and MPK/phenol(J,K)
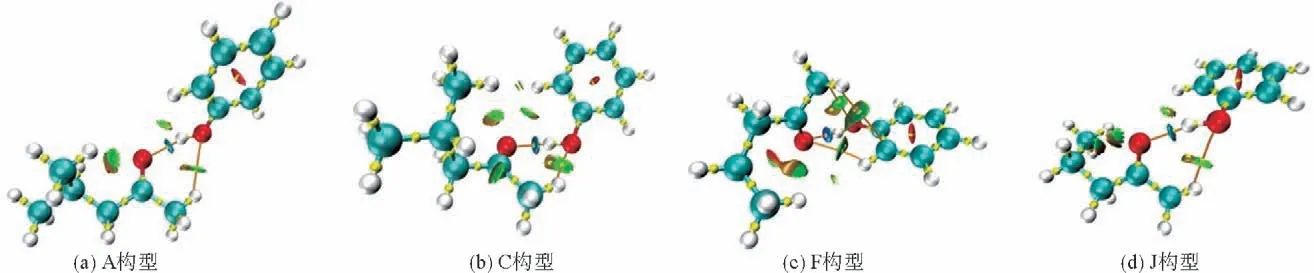
图4 具有临界点和键路径的MIBK/苯酚(A、C)和MPK/苯酚(F、J)的RDG图Fig.4 RDG graphs of MIBK/phenol(A,C)and MPK/phenol(F,J)with critical points and bond paths
从表8可以得出,4种构型中相互作用能都是由OH—O 氢键主导,萃取剂的萃取能力的大小由该氢键键能主导,同时图4中的键临界点和键路径确定了氢键的存在。
同时对比构型A、C 和F、J,在O31 H32-O19(O26 H27-O14)氢键起主导作用的条件下,C(F)比A(J)氢键键能低,氢键强度大,直接作为C(F)比A(J)相互作用能低的主要原因。与此同时F 构型的氢键数量最多,氢键强度最大,萃取效果最好,即MPK 的萃取效果大于MIBK 的萃取效果,验证模拟结果。
3 结 论
1)对煤气化反应工段、脱酸工段、萃取脱酚工段进行全流程模拟,并得到两种萃取剂对废水中酚类萃取效果的对比。
2)萃取剂单体的不同构象会影响团簇的稳定性和相互作用能的大小,进而影响萃取效果。能量越低,构型越稳定;相互作用能越低,萃取效果越好。
3)分子间氢键在团簇中起到至关重要的作用。对于本工作涉及到的几种团簇,酚羟基与酮羰基形成的氢键起主导作用,同时影响相互作用能大小,氢键键能越低,相互作用能越低,体系越稳定,萃取效果越好。
4)MPK/苯酚的J构型在同一团簇中能量最低,结构最稳定;在不同团簇中,氢键数量最多,强度最高,萃取效果最好。总之,MPK 的处理效果要高于MIBK,验证Aspen模拟结果。
猜你喜欢
中国化肥信息(2022年9期)2022-11-23
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
选煤技术(2022年3期)2022-08-20
矿业科学学报(2021年6期)2021-11-06
波谱学杂志(2021年3期)2021-09-07
科学技术与工程(2021年16期)2021-07-12
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
科学与信息化(2020年28期)2020-12-21
山东工业技术(2018年15期)2018-09-26
科学中国人(2018年15期)2018-01-29
