
中枢神经细胞瘤临床病理观察及文献复习
2021-01-29 03:20刘雅玲朱含梅刘银华李佳嘉徐五琴汪向明
皖南医学院学报 2021年1期
刘雅玲,朱含梅,刘银华,李佳嘉,徐五琴,于 涛,汪向明
(皖南医学院 研究生学院,安徽 芜湖 241002;2.皖南医学院第一附属医院 弋矶山医院 a.病理科;b.重症医学科,安徽 芜湖 241001)
1982年Hassoun等首次提出中枢神经细胞瘤(central neurocytoma,CN)的病理学诊断概念[1],CN是中枢神经系统少见的肿瘤,约占颅内肿瘤的0.1%~0.5%[2],研究发现CN起源于残余原始神经上皮组织,是生长于第三脑室与侧脑室的小细胞神经元肿瘤,其最主要的发生部位在透明隔近室间孔处(Monro孔),有文献报道发生于第四脑室的病例[3]。其主要体征是梗阻性脑积水及颅内压增高引起的头痛,事实上某些CN患者早期可以出现一些高级神经功能障碍的表现,如记忆力下降、认知功能减退[4]。在2016年WHO中枢神经系统肿瘤分类中,CN被归类为神经元和混合性神经元胶质肿瘤,WHO分级为Ⅱ级;一般认为其预后较好[5]。
1 资料与方法
1.1 一般资料 2016年1月~2019年2月弋矶山医院住院CN患者3例。患者年龄18~42岁,平均年龄(27±9)岁;所有病例均行CT、MRI检查,并经病理学检查确诊。
1.2 治疗 2例患者行额部开颅经纵裂肼胝体入路侧脑室肿瘤显微切除术,1例患者行脑室内肿瘤切除术。
1.3 病理检查方法
1.3.1 HE制片观察 所有标本均经10%中性缓冲福尔马林溶液固定,石蜡包埋,4 μm切片,常规苏木精-伊红染色法(HE)染色。
1.3.2 免疫组织化学检查 检测肿瘤组织中相关抗体的表达情况:广谱细胞角蛋白(CKpan)、上皮膜抗原(EMA)、突触素(Syn)、嗜铬素A(CgA)、胶质纤维酸性蛋白(GFAP)、人神经丝蛋白(NF)、髓鞘相关糖蛋白(CD57)、神经特异性烯醇(NSE)、Ki-67、酸性钙结合蛋白(S-100)、神经元特异核蛋白(NeuN)等,免疫组化染色按说明书操作。
1.3.3 试剂 鼠抗人单克隆抗体(CKpan、EMA、Syn、CgA、GFAP、NF、CD57、NSE及NeuN)均购自福州迈新生物技术开发有限公司。
2 结果
2.1 临床资料 患者1,女性,18岁;首发症状:突发一过性意识丧失5 d余,持续2 s(短暂性失忆),30 min后恢复,送医过程中呕吐两次;神经专科检查显示神志清楚,双侧瞳孔等大等圆,无病理性反射;头颅MRI平扫+增强提示:左侧侧脑室旁孟氏孔-三脑室结节灶,左侧侧脑室扩大积水。患者2,男性,23岁;首发症状:体检发现右侧侧脑室占位1个月余;神经专科检查显示神志清楚,双侧瞳孔等大等圆,无病理性反射;于外院行头颅MRI平扫+增强提示:右侧侧脑室体部占位。患者3,男性,42岁;首发症状:双眼视力下降半年余,间断头痛、呕吐2周;神经专科检查显示神志清楚,双侧瞳孔等大等圆,无病理性反射;于外院行头颅MRI平扫+增强提示:鞍上区至两侧脑室间占位,并脑积水(图1)。
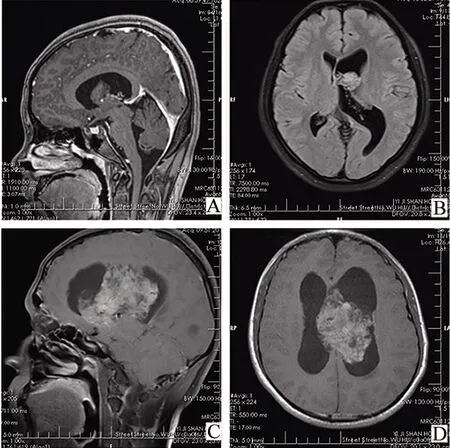
A、B.患者1,左侧脑室近孟氏孔处见一直径约2.0 cm信号影,信号略欠均,边界尚清,增强后呈轻度强化,双侧脑室扩张;C、D.患者3,左侧脑室内见团块状不均匀较明显强化影,边缘散在斑片状、结节状低强化区,呈“皂泡样”表现,边界不清,上缘以宽基底与侧脑室顶壁相贴,病灶局部突入右侧室,下方压迫第三脑室。
2.2 组织大体检查 患者1术中切开脑室壁间肿瘤组织,质地较软,色灰白,血供一般,肿瘤基底位于室管膜,粘连紧密,体积约2.0 cm×1.5 cm×1.5 cm;患者2术中切开脑室壁即见肿瘤组织,质地较软,色灰白,血供丰富,肿瘤基底位于室管膜,粘连紧密,体积约3.5 cm×2.0 cm×1.5 cm;患者3术中探查脑室即见肿瘤组织,肿瘤呈鲜红色,质地稍硬,血供丰富,体积约7.0 cm×5.0 cm×5.0 cm,肿瘤自胼胝体膝部向前及下方生长,与透明隔无明显界限,室间孔被肿瘤组织阻塞。
2.3 病理结果 光镜下:3例患者瘤细胞均呈弥漫大小一致,核呈圆形或卵圆形,部分区域见核周空晕,细胞质均染,呈透明或淡红色,核仁不明显,核分裂象罕见,瘤细胞周血管呈分枝状;其中1例可见无核神经岛结构,瘤细胞穿入其中;1例见灶区瘤细胞排列呈流水状,瘤细胞围绕血管排列呈围血管的菊形团[7-9]。免疫组化分析:3例Syn、NSE、NeuN、GFAP、CD57、S-100蛋白均阳性,Ki-67(1%~3%,+)、CKpan、EMA、NF、CgA均阴性(图2)。
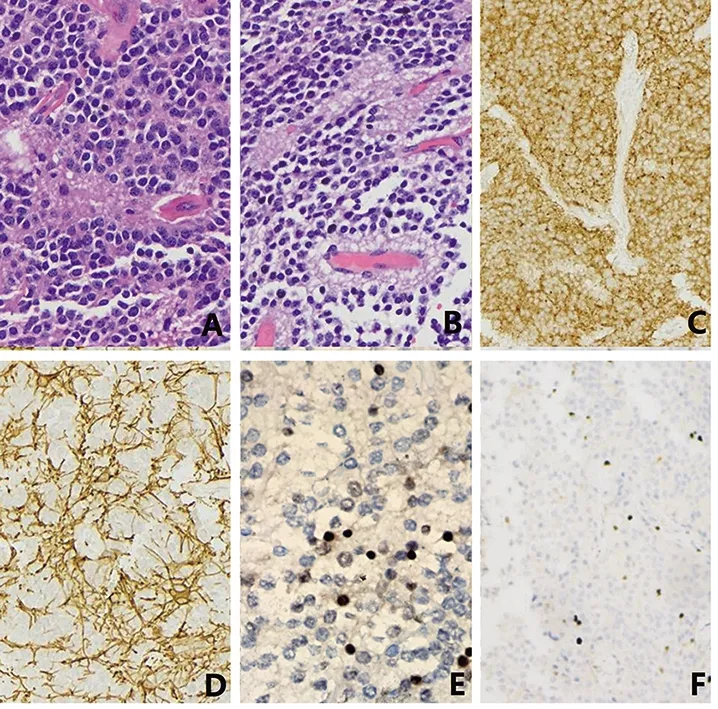
A.可见瘤细胞大小一致,核圆胞质均染,可见无核神经纤维岛(HE,×200);B.可见瘤细胞围绕血管形成菊形团(HE,×200);C.瘤细胞神经突触素Syn弥漫阳性(×200);D.瘤细胞NSE弥漫阳性(×200);E.部分瘤细胞细胞核表达NeuN(×200);F.瘤细胞Ki-67(1%阳性)(×200)。
2.4 随访 3例均获随访,以手术日期定为生存日期的开始,随访日期截至到2020年1月,随访时间11~44个月,中位随访时间27个月,随访期间无复发或转移。
3 讨论
3.1 临床表现 CN以中青年人常见,多发于20~40岁,1~80岁均可发病,男女发病比例相同[9],本组发病年龄18~42岁,男1例,女2例。CN典型发病部位是在侧脑室或第三脑室处,本组3例患者均发生于侧脑室。CN通常病史短,患者会出现颅内压增高症状,部分可出现高级神经功能障碍,偶见视力和智力障碍;位于隔区、第三脑室和下丘脑的CN可引起内分泌紊乱,本组仅1例发生一过性意识丧失,其余2例均体检发现,一定程度上说明CN呈慢性病程,患者无明显的临床表现。影像学方面,肿瘤在CT上表现为均匀的等密度或稍高密度影,增强后可强化,可见钙化和“皂泡样”和“丝瓜瓤样”囊性变[10-11]。
3.2 组织病理学特点 本组3例瘤细胞弥漫分布,大小一致,细胞核圆形或椭圆形,染色质均匀,部分见核周空晕,1例见无核神经岛,1例见血管周假菊形团,与文献相符;若病例中存在坏死、血管内皮增生、核异型或高核分裂活性(>3个/10 HPF),以及增殖指数增高(>3%),称为非典型中枢神经细胞瘤,提示肿瘤易复发或预后较差[8,11]。
3.3 免疫表型 Syn是CN的主要分子标记之一,其他标记物包括NSE、NeuN,HNK1(Leu7)亦有表达,GFAP不表达。本组病例中3例Syn、NSE、NeuN均呈阳性表达。
3.4 CN的分子遗传学 传统的细胞学遗传学分析显示CN有较为少见的染色体异常。有文献[12-13]报道l例中枢神经细胞瘤中具有17号染色体的缺失,部分文献报道应用荧光原位杂交技术发现9例中存在3例有7号染色体的获得。有文献研究10例原发性CN,60%的CN显示出染色体的失衡,在染色体2 p、10 q和18 q上都发现了较高比例的遗传物质的获得,40%的中枢神经细胞瘤显示出染色体2 p 22至P远端遗传物质的获得,可能在该区域存在与CN发病有关的癌基因。
3.5 CN的鉴别诊断
3.5.1 少突胶质细胞瘤 少突胶质细胞瘤与CN细胞组织学特点相似,瘤细胞弥漫排列,形态较单一,核圆,核周有“空晕”,部分少突胶质细胞瘤局灶表达Syn,但多不表达NSE及NeuN。NeuN、Olig2、Syn和NSE可用来鉴别中枢神经细胞瘤和少突胶质细胞瘤[14-15];现有研究发现分子特征IDH1/2基因突变和1p/19q染色体臂共缺失是其诊断的要点。
3.5.2 透明细胞型室管膜瘤 透明细胞型室管膜瘤是一种罕见的脑肿瘤[16-17],瘤细胞界限清楚,胞浆透亮;CN与透明细胞型室管膜瘤均可钙化或形成假菊形团,免疫组化显示GFAP与EMA阳性,Syn及NeuN阴性;神经丝蛋白和神经元在大多数透明细胞型室管膜瘤中表达,而CN则相反,且常出现无核神经岛。
3.5.3 胚胎发育不良性神经上皮瘤(dysembryoplastic neuroepithelialtumor,DNT) DNT是一种较少见的神经元和神经胶质细胞混合性肿瘤,相当于WHO Ⅰ级。常见于儿童、青少年,可引起癫痫发作,肿瘤主要特征是定位于皮质内和呈结节状[18],由特异性胶质神经元成分混合而成,包括少突胶质细胞样细胞、星形细胞和漂浮正常的神经元细胞,各成分比例不一,主要以少突胶质细胞样细胞为主;DNT病变通常与局灶性皮质发育异常有关[19]。免疫组化标记显示瘤细胞表达NF、Syn、S-100、GFAP。
3.6 治疗及预后 CN目前以手术切除为主,手术切除肿瘤需根据肿瘤位置评估,可经侧脑室额中回入路或侧脑室三角区入路切除肿瘤,本组3例均经纵裂肼胝体入路侧脑室肿瘤显微切除,术中需根据肿瘤与侧脑室壁的关系决定行肿瘤全切除或近全切除,特别需要注意肿瘤与第三脑室之间关系;如肿瘤与第三脑室关闭,可以放弃手术全切肿瘤,可近全切,术后再加做放射治疗增加疗效,也有文献提出立体定向放射外科手术作为辅助治疗手段,通常是针对异常恶性的形式和病例残余大的肿瘤病灶[20]。
猜你喜欢
昆明医科大学学报(2021年5期)2021-07-22
中华养生保健(2021年18期)2021-02-13
中国临床医学影像杂志(2019年4期)2019-06-18
中国临床医学影像杂志(2019年2期)2019-04-25
中国妇幼健康研究(2019年2期)2019-03-26
影像研究与医学应用(2019年7期)2019-03-20
磁共振成像(2015年1期)2015-12-23
郑州大学学报(医学版)(2015年2期)2015-02-27
中国实用医药(2015年36期)2015-02-01
中国卫生标准管理(2015年13期)2015-01-26
