
金黄色葡萄球菌杀白细胞素基因敲除菌株的构建及生长特性分析
2021-01-22 02:57刘文宇王长珍王凌利刘思国周学章张万江
中国预防兽医学报 2020年11期
刘文宇,祝 瑶,王长珍,杨 亲,栾 天,王凌利,刘思国,周学章,张万江*
(1.中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室/动物细菌病创新团队,黑龙江 哈尔滨 150069;2.宁夏大学生命科学学院,宁夏 银川 750021)
金黄色葡萄球菌(Staphylococcus aureus)是一种引起人和动物感染的重要致病菌,能够引起皮肤、肺等局部感染以及败血症、脓毒症等全身性感染性疾病,严重可致死亡,威胁着人类和动物健康[1]。金黄色葡萄球菌能够产生多种与致病性相关的细胞毒素,主要包括γ-溶血素和杀白细胞素(Panton-valen⁃tine leukocidin,PVL)。其中,PVL 是穿孔毒素,能够引起人体内白细胞的细胞毒性变化[2],可致白细胞溶解和组织坏死。PVL 参与金黄色葡萄球菌引起的原发性皮肤感染、坏死性肺炎、肌肉骨骼损伤[3],可作为其引起的侵袭性疾病的流行病学调查指标,例如菌血症、骨骼肌肉疾病以及软组织感染等[4]。此外,研究还发现PVL 是耐甲氧西林金黄色葡萄球菌(MRSA)的主要毒力因子[5]。PVL 由LukSPV 和LukF-PV 这两个共转录基因组成,二者编码的相应多肽组装在一起构成具有活性的PVL 蛋白[3]。尽管目前研究表明PVL 在金黄色葡萄球菌致病性中发挥着重要作用,但该毒力因子具体的致病机理还不十分明确,本实验以金黄色葡萄球菌ATCC 49775株为研究对象,利用同源重组的方法构建PVL 基因敲除菌株,并对敲除菌株的稳定性和生长特性进行评价,为进一步阐明金黄色葡萄球菌PVL 的致病机理奠定基础。
1 材料与方法
1.1 主要实验材料金黄色葡萄球菌ATCC 49775株、RN4220 株、质粒pBT2、pLI50、pEC1 均由中国农业科学院哈尔滨兽医研究所动物细菌病研究团队保存;E. coli DH5α 感受态细胞和细菌基因组DNA提取试剂盒购自天根生化科技(北京)有限公司;溶葡萄球菌素购自Amresco 公司;溶菌酶购自Coolaber公司;红霉素购自BBI 公司;氯霉素购自Amresco 公司;质粒提取试剂盒和DNA 回收试剂盒购自Omega公司;PCR 试剂、T4 DNA 连接酶和限制性内切酶均购自TaKaRa 公司。
1.2 引物设计与合成参考文献[6],根据GenBank中金黄色葡萄球菌PVL 基因序列(NC_002321),设计扩增PVL 基因上、下游引物以及鉴定引物。根据质粒pEC1 中红霉素抗性基因ermB 的序列设计引物(表1)。引物均由吉林库美生物科技有限公司合成。

表1 PCR 引物序列Table 1 Primers used in this study
1.3 敲除质粒和回补质粒的构建与鉴定以提取的金黄色葡萄球菌ATCC 49775 基因组DNA 为模板,以up-0706-1-F/ up-0706-1-R 和down-0706-3-F/down-0706-3-R 为引物PCR 扩增PVL 基因上下游同源臂;以质粒pEC1 为模板,以0706-2-F/0706-2-R引物,PCR 扩增红霉素抗性基因ermB 片段;以上述片段为模板,up-0706-1-F/down-0706-3-R 为引物通过融合PCR 的方法扩增上游片段-红霉素抗性基因-下游片段,克隆至pBT2 载体中,提取重组质粒,利用引物up-0706-1-F/up-0706-1-R 经PCR 鉴定,并经BamHI 酶切鉴定后测序鉴定,获得重组质粒命名为pBT2-ΔPVL。以金黄色葡萄球菌ATCC 49775 基因组为模板,0905-2-F/ 0905-2-R 为引物扩增回补序列克隆至pLI50 穿梭质粒后,转化至大肠杆菌DH5α 感受态,筛选阳性克隆获得重组质粒,酶切并测序鉴定,获得的回补质粒命名为pLI50-PVL。
1.4 PVL 基因敲除菌株和回补菌株的筛选与鉴定将构建的敲除质粒pBT2-ΔPVL 电转化至金黄色葡萄球菌RN4220,经RN4220 修饰后再以相同条件转化至金黄色葡萄球菌ATCC 49775 中,利用引物up-0706-1-F/down-0706-3-R 经PCR 和测序鉴定,阳性菌株命名为ΔPVL。将阳性菌株接种于红霉素抗性(2.5 μg/mL)的TSB 液体培养基中,30 ℃培养6 h 后转至42 ℃培养24 h。再涂布于红霉素抗性(2.5 μg/mL)的TSA 琼脂板中,30 ℃培养12 h 后,挑取单克隆接种于红霉素抗性(2.5 μg/mL)的TSB 液体培养基,30 ℃培养12 h 后,转接至氯霉素抗性(15 μg/mL)的TSB 液体培养基,30 ℃继续培养12 h,筛选具有红霉素抗性但氯霉素抗性缺失的菌株。提取该菌株的基因组,通过鉴定引物0907-1-F/0907-1-R 经PCR鉴定。根据以上方法,将回补质粒pLI50-PVL 电转化至金黄色葡萄球菌RN4220 进行修饰,修饰后的重组质粒电转化至敲除菌株ΔPVL,构建敲除菌株的回补菌株,命名为CΔPVL。提取CΔPVL 基因组,通过鉴定引物0905-2-F/0905-2-R经PCR鉴定。根据以上方法,将回补质粒pLI50-PVL 电转化至金黄色葡萄球菌RN4220 进行修饰,修饰后的重组质粒电转化至敲除菌株ΔPVL,构建敲除菌株的回补菌株,命名为CΔPVL。提取CΔPVL 基因组,通过鉴定引物0905-2-F/ 0905-2-R 经PCR 鉴定。
1.5 敲除菌株体外遗传稳定性的检测将敲除菌株ΔPVL 在TSB 培养基中连续传代培养,分别提取第5 代、10 代、15 代、20 代、25 代、30 代 的 菌 液 基 因组DNA,利用鉴定引物0907-1-F/0907-1-R 经PCR鉴定,检测敲除菌株在体外的遗传稳定性。
1.6 敲除菌株和回补菌株体外生长曲线的测定将敲除菌株ΔPVL、回补菌株CΔPVL 与金黄色葡萄球菌ATCC 49775 37 ℃在TSB 液体培养基中,37 ℃220 r/min 培养,每隔30 min 取200 μL 菌液测定一次OD600nm值[7],试验重复3次,绘制3种菌株的生长曲线。
1.7 统计分析所有数据均采用SPSS 17.0 统计软件进行分析,p<0.05 表示差异显著。
2 结 果
2.1 敲除质粒和回补质粒的构建与鉴定结果通过PCR 方法扩增获得PVL 基因的同源臂上、下游片段、红霉素抗性基因ermB 片段,以上述3 个扩增片段为模板,经融合PCR 扩增获得2 542 bp 融合片段(图1A)。将融合片段克隆至质粒pBT2 中构建重组质粒pBT2-ΔPVL,PCR和鉴定结果显示,在约500 bp出现目的条带(图1B),测序结果显示克隆至pBT2 的融合片段与同源臂上游片段-红霉素抗性基因-同源臂下游片段的序列一致,表明正确构建敲除质粒pBT2-ΔPVL。将PCR 扩增获得的PVL 基因克隆至pLI50 质粒获得PVL 全基因回补质粒pLI50-PVL,酶切鉴定(图2)、测序结果均正确,表明正确构建回补质粒pLI50-PVL。
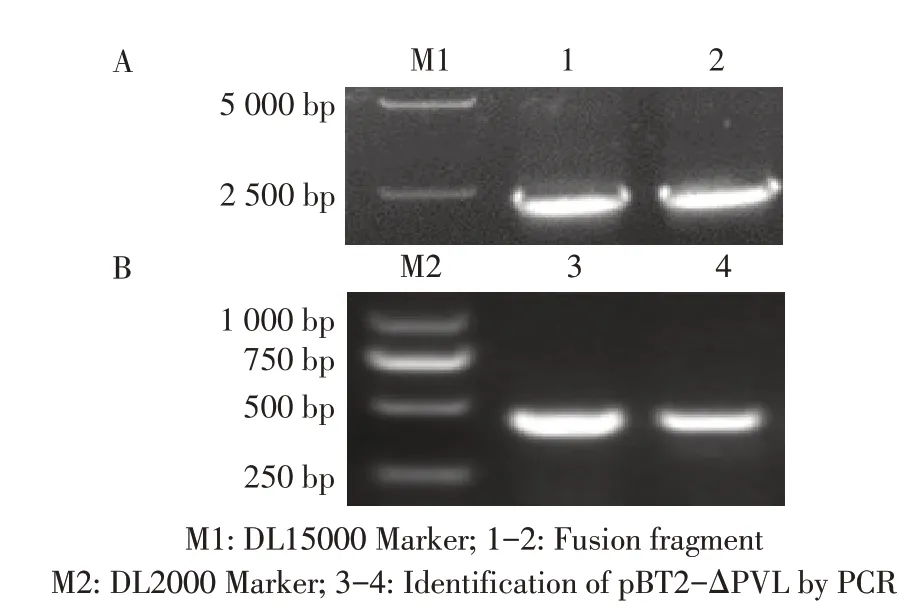
图1 3 个基因片段融合PCR 扩增(A)及敲除质粒的PCR(B)鉴定结果Fig. 1 Fusion PCR amplification of 3 gene fragments (A) and PCR identification results of knockout recombinant plasmids (B)

图2 回补质粒的酶切鉴定结果Fig. 2 Identificationof the replenishment plasmid by digestion
2.2ΔPVL 菌株的构建、筛选和鉴定结果将在金黄色葡萄球菌RN4220 中修饰后的敲除质粒pBT2-ΔPVL 电转化至金黄色葡萄球菌ATCC 49775,于30 ℃和42 ℃交替多次传代以及抗性反复筛选,提取疑似阳性克隆菌株的基因组DNA,利用鉴定引物0907-1-F/0907-1-R 经进行PCR 鉴定。结果显示,敲除菌株ΔPVL 基因组DNA 扩增出2 500 bp 的目的片段,而野生金黄色葡萄球菌ATCC 49775 基因组DNA 扩增的片段为3 110 bp(图3A),表明正确构建了敲除PVL 基因的金黄色葡萄球菌ΔPVL 株。将pLI50-PVL 转入敲除菌株ΔPVL 内,获得回补菌株CΔPVL。利用0905-2-F/0905-2-R 进行PCR 鉴定,CΔPVL 扩增得到与野生菌株大小一致的目的条带(图3B);扩增产物测序结果证实回补菌株CΔPVL内含有正确的PVL 基因。以上结果表明获得敲除菌株ΔPVL 的回补菌株CΔPVL。
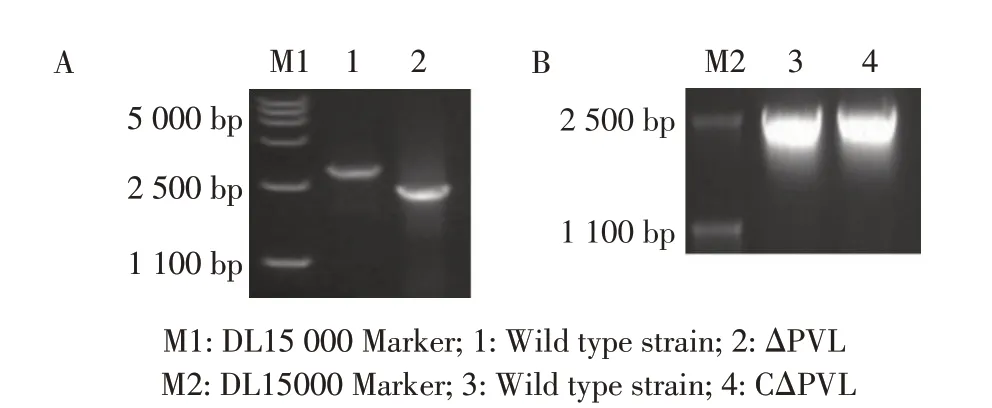
图3 敲除菌株ΔPVL(A)和回补菌株CΔPVL(B)的PCR鉴定结果Fig. 3 Identification of deletion mutant ΔPVL(A) and supplementary strain CΔPVL(B) by PCR
2.3 敲除菌株的体外遗传稳定性检测结果对敲除菌株ΔPVL 进行体外连续传代,提取第5、10、15、20、25、30 代菌液,利用鉴定引物0907-1-F/0907-1-R 进行PCR 鉴定。结果显示,各代次敲除菌株ΔPVL 均能扩增出约2 500 bp 的目的条带(图4)。表明敲除菌株连续传代未发生回复突变,敲除菌株能够在体外稳定遗传。
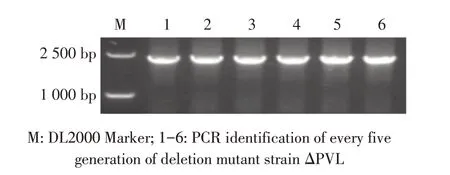
图4 敲除菌株ΔPVL 遗传稳定性PCR 鉴定结果Fig. 4 Identification of genetic stability of deletion mutant ΔPVL by PCR
2.4 敲除菌株和回补菌株体外生长曲线的测定在相同条件下分别培养敲除菌株ΔPVL、回补菌株CΔPVL 与金黄色葡萄球菌ATCC 49775(WT),根据OD600nm值绘制生长曲线。结果显示,ΔPVL 在体外生长较慢并且其生长速率显著低于ATCC 49775 野生菌,在培养2 h~4.5 h 生长速率差异显著,CΔPVL 在体外生长速率与ATCC 49775 野生菌基本一致(图5)(p<0.01)。表明,PVL 基因的缺失可能影响了金黄色葡萄球菌ATCC 49775 的生长速率,推测PVL 的合成与金黄色葡萄球菌的生长性能相关。

图5 敲除菌株ΔPVL、回补菌株CΔPVL 与金黄色葡萄球菌ATCC 49775 生长速率的比较Fig. 5 Comparison of growth rates of deletion mutant ΔPVL,the replenishing strain CΔPVL and S. aureus ATCC 49775
3 讨 论
PVL 由LukS-PV 和LukF-PV 这两个共转录基因组成,其首次在金黄色葡萄球菌ATCC 49775 中被发现,是一种对人类和兔子多核型白细胞具有高度特异性的白细胞素[8]。PVL 作为金黄色葡萄球菌的毒力因子,在金黄色葡萄球菌中并不常见,因此对于PVL 的潜在威胁知之甚少[4]。目前已有研究报道PVL与原发性皮肤感染和肺炎有关[3],并且对其在皮肤、大脑、呼吸系统和肌肉骨骼等器官或系统的研究结果显示,PVL 逐渐成为一种新的全球公共卫生威胁[9-11]。
在对病原微生物的研究中,靶基因的置换或缺失是确定该基因功能、阐明其致病机理的重要手段[12]。目前,利用同源重组的方法对目标基因敲除是研究细菌单基因功能的常用方法,在革兰氏阴性菌中广泛应用[13]。
本研究分别以温敏型质粒pBT2 和穿梭质粒pLI50 为载体,利用同源重组的方法构建了金黄色葡萄球菌ATCC 49775 PVL 的基因敲除菌株和该基因的回补菌株。在构建的敲除质粒中引入ermB 基因作为抗性筛选标记,使其具有红霉素抗性,同源重组后一方面将红霉素抗性基因引入ATCC 49775 野生菌株中,使重组菌株获得红霉素抗性,进而利用红霉素筛选阳性重组菌;另一方面,ermB 基因整合替换了野生菌株中的PVL 基因,进而完成了对PVL 基因的敲除。
此外,通过对敲除菌株ΔPVL 和回补菌株CΔPVL 体外生长特性的测定表明ΔPVL 与野生型ATCC 49775 菌株相比生长速率显著下降,回补菌株CΔPVL 与野生型ATCC 49775 菌株生长速率基本一致,目前还无PVL 基因与金黄色葡萄球菌的生长代谢相关的研究报道。本研究表明,PVL 基因可能与金黄色葡萄球菌的生长调节有关,具体机制还需要进一步研究。
本研究构建的金黄色葡萄球菌ATCC 49775 的敲除菌株,为进一步研究PVL 基因的功能及其在金黄色葡萄球菌感染中的致病机理奠定了基础。
猜你喜欢
保健与生活(2022年7期)2022-04-08
华声文萃(2021年11期)2021-11-15
文萃报·周五版(2021年36期)2021-09-16
食品安全导刊(2021年20期)2021-08-30
中华养生保健(2020年3期)2020-11-16
科研成果与传播(2019年3期)2019-09-10
创新作文(1-2年级)(2018年2期)2018-09-13
学习报·教育研究(2017年33期)2017-08-31
家庭科学·新健康(2014年10期)2014-10-24
食品工业科技(2014年23期)2014-03-11
