
早期生长反应蛋白1参与肝病发生主要病理机制研究进展
2021-01-22 02:28:58曾伟兰
中国药理学与毒理学杂志 2020年9期
曾伟兰 ,汪 艳
(南方医科大学1.药学院,2.中心实验室,广东 广州 510515;3.广东省肝纤维化工程技术研究中心,广东 广州 510515)
早期生长反应蛋白1(early growth response factor protein 1,EGR1)属于即刻早期基因家族成员之一,是参与调控细胞生长、分化以及凋亡的重要转录因子[1],在肝、心、脑、肺、脾、骨骼肌、肾、卵巢和前列腺等组织均可检测到其表达[2],但在大多数正常组织的表达水平较低[1-3]。EGR1在转化生长因子β1(transforming growth factor-β1,TGF-β1)、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)、肝细胞核因子 4α(hepatocyte nuclear factor 4α,HNF4α)、转录因子E2F1(E2F transcription factor 1,E2F1)以及小异二聚体伴侣(small heterodimer partner,SHP)等调控分子的作用下,通过多种机制参与肝再生、肝纤维化、酒精性脂肪肝和肝细胞癌等生理和病理进程。现有综述文献主要集中于EGR1与各种肿瘤性疾病的相关性,关于EGR1参与调控肝生理病理等过程的系统综述尚较少报道。本文针对EGR1主要生物学功能及其参与肝病理发生机制等研究进展进行综述,并讨论其潜在的临床意义。
1 EGR1分子的结构特点与功能调控
EGR1也称为Zif268,NGFI-A,Krox24或TIS8等,是一种Cys2-His2型核转录因子,普遍存在于从酵母到人类的真核细胞。人类EGR1编码基因位于染色体5q23-q31上。EGR1蛋白分子含543个氨基酸残基,包括锌指DNA结合结构域、反式激活结构域、NGFI-A结合蛋白(NGFI-A binding protein,NAB)相互作用结构域及核定位序列。NAB1和NAB2相互作用结构域,可抑制EGR1的转录活性[4-5]。NAB2的表达本身可以由EGR1控制,当EGR1过表达时,EGR1的锌指结构识别NAB2和NAB1结合位点,而NAB结构域使EGR1无法识别特异DNA序列,抑制EGR1转录调控过程,最终对EGR1表达水平发生负反馈调节作用[5]。因此,EGR1能够通过负反馈调节机制调节自身的活性水平。
人EGR1基因启动子含有5个血清反应元件,可被血清反应因子及三元复合因子(ternary complex factor)所识别。EGR1激活受MAPK家族调控,包括细胞外信号调节蛋白激酶(extracellular signalregulated protein kinase,ERK)、c-Jun NH2-末端激酶和P38 MAPK。EGR1是MAPK家族信号通路的下游靶点。磷酸化的ERK由细胞质转运至细胞核可直接激活EGR1,使其参与细胞信号相关的生理调控[6]。
与其他即刻早期基因一样,由于激动剂性质和细胞类型的不同,EGR1动态转录在诱导时间方面有所差异[7]。EGR1的表达与环境性刺激因素有关,包括生长因子类、细胞因子类、激素、凝血酶、剪切应力和机械力、神经递质、紫外线以及缺氧等[8-11],其活性调控的修饰途径包括乙酰化的负调控、磷酸化以及泛素化的正调控[12-13]。
2 EGR1的下游靶基因信号通路及其分子机制
EGR1的锌指结构能高度识别富含G-C的核苷酸序列,以锌指依赖方式激活或抑制相关靶基因的转录。EGR1下游靶基因主要涉及了参与细胞生长或分化、凋亡和应激反应等过程的基因,如与细胞周期停滞和凋亡相关的第10号染色体缺失的磷酸酶和张力蛋白同源物基因(phosphatase and tensin homologue deleted on chromosome ten,PTEN),P53,P21,P73及Bcl-2家族的促凋亡蛋白等[14-15]。此外,EGR1不仅调控炎症介质分子的表达,如细胞间黏附分子1、单核细胞趋化蛋白1、巨噬细胞炎症蛋白2等;还调控纤维化发生相关分子的表达,如促纤维化细胞因子、血小板衍生生长因子、结缔组织生长因子和血管内皮生长因子等[16-17]。在调控肿瘤细胞功能方面,EGR1通过调节各种生长因子、抑癌基因、细胞周期蛋白或凋亡相关因子,介导细胞生长和肿瘤的发生,如EGR1直接诱导的PTEN可抑制肝癌细胞的迁移与侵袭[18],活性氧(reactive oxygen species,ROS)激活的 EGR1可经 ROS/MAPK/EGR1/Bax信号通路与靶基因Bax结合,诱导肾上腺皮质癌细胞凋亡[19]。综上,EGR1调控的靶基因种类丰富,提示EGR1具有重要的生物学功能。
3 EGR1的主要生物学功能
3.1 肝生理学功能
3.1.1 肝再生
肝具有强大的再生能力。在大鼠模型,正常肝进行部分肝叶切除后,在术后5~9 d即可恢复原肝体积。研究发现[20],肝切除1/3术后48和72 h,EGR1表达水平在伤口相邻区域最高,提示EGR1可能参与肝损伤修复过程。EGR1在肝再生起始阶段被显著诱导,并调控细胞周期相关分子表达,如细胞周期蛋白和细胞分裂周期蛋白20等[21-22]。如用腺病毒特异性地抑制肝EGR1的转录,发现细胞周期蛋白D1和E表达水平均降低,香叶基香叶基二磷酸合酶(geranylgeranyl diphosphate synthase,GGPPS)和下游RAS/MAPK途径的活性均受到抑制[22]。故EGR1可能通过激活GGPPS/RAS/MAPK下游通路,或促进G1/S期细胞周期蛋白的表达以调节肝再生过程。
然而,EGR1相关肝再生作用在肝毒素模型与部分肝切除模型可能存在差异。如果敲除EGR1,CCl4介导的肝损伤会发生G1/S期阻滞,而部分肝切除的小鼠表现为G2/M期阻滞[21]。P38 MAPK可在G2期促进细胞周期进程。EGR1敲除的部分肝切除模型表现出P38 MAPK异常磷酸化,故可能是引起G2/M期阻滞的部分原因。另外,不同肝损伤模型之间线粒体状态的差异,细胞能量状态和氧化还原状态不同,也可能会影响到不同的细胞周期进程[23]。CCl4诱导的EGR1敲除小鼠的肝损伤模型抗氧化功能比部分肝切除的小鼠弱,故CCl4肝损伤模型可能延迟了细胞周期阶段DNA的合成,最后造成该差异[23]。
值得注意的是,在肝再生过程中,调控EGR1表达的特异性分子机制仍尚不清楚。细胞外ATP[24]和P2Y2嘌呤能受体[25]等可能有重要作用。但至今相对明确的是,EGR1可通过调控细胞周期相关分子的表达,提高肝细胞存活,促进肝再生进程。EGR1也许可以作为肝再生过程的新治疗靶标。
3.1.2 肝胆固醇合成代谢
EGR1是胆固醇合成相关基因的直接调控因子,如3-羟基-3-甲基戊二酰-CoA还原酶(3-hydroxy-3-methylglutaryl-CoA reductase,HMGCR)和角鲨烯环氧酶等。HMGCR活性是胆固醇合成的限速步骤。在HMGCR启动子-130位点具有一段富含GC的EGR1结合序列,EGR1与该序列结合可促进HMGCR的转录[26],提示EGR1与胆固醇合成存在密切联系。另外,生物信息学分析显示,EGR1可激活生长因子来诱导胆固醇生物合成。EGR1可与甾醇调节元件结合蛋白2(sterol regulatory element binding protein-2,SREBP-2)发生协同作用,介导胰岛素诱导肝胆固醇的合成[26-27]。综上,EGR1可能是肝调节胆固醇生物合成途径的手段之一。
3.2 细胞的生长、分化和凋亡
EGR1的生物学功能一般涉及胚胎发育、细胞生长、分化和凋亡等。EGR1可能在小鼠胚胎植入过程刺激环氧合酶2和血管内皮生长因子等基因的表达,促进新血管生成,参与胚胎发育过程[28]。在细胞周期的G0/G1期,EGR1发挥调节细胞生长和分化的作用。在部分肝切除的野生型小鼠中,EGR1 mRNA表达水平在肝细胞进入G0/G1期时显著增加;敲除EGR1小鼠肝细胞有丝分裂进程发生延迟[21]。另外,EGR1在新生小鼠腹侧的表达处于高水平;当小鼠生长至21 d,EGR1水平降低至基线[20]。由此提示,EGR1高表达主要集中在细胞生长增殖活跃时期,对细胞的生长增殖可能具有促进作用。据报道,EGR1通过激活Wnt/β联蛋白下游通路的支架蛋白,参与小鼠小肠上皮细胞的分化[29]。此外,EGR1可以线粒体依赖性方式,经NF-κB下游信号通路促进卵泡闭锁的颗粒细胞凋亡[30]。
3.3 肝纤维化
肝纤维化是细胞外基质的弥漫性过度沉积及瘢痕形成[31]。肌成纤维细胞通过促进胶原合成、分泌以及细胞外基质累积过程是纤维化形成的基础。肝星状细胞(hepatic stellate cells,HSC)是肝肌成纤维细胞的重要来源,数量约占所有肝细胞的15%,是参与肝纤维化的主要细胞类型。
HSC通常处于“静止”状态。TGF-β1可促使HSC转分化成肌纤维细胞,并使其合成过量的基质蛋白(如纤连蛋白、胶原蛋白和弹性蛋白等),促进肝纤维化的发生[32]。对乙酰氨基酚低剂量(200 mg·kg-1)造模6周的EGR1敲除小鼠,HSC活化、胶原蛋白以及TGF-β1表达水平均增强[33]。但另有报道,EGR1可增加体外培养HSC激活标志物的表达,如α-平滑肌肌动蛋白和Ⅰ型胶原蛋白[34]。目前导致EGR1对HSC活化具有不同表现的原因尚不清楚。
肝细胞通过上皮-间充质转化(epithelial-mesenchymal transition,EMT)可促进成纤维细胞活化[35]。ELK3(ETS致癌基因家族成员)可促进肝纤维化EMT过程[36]。EGR1是组织损伤诱导下ELK3的下游靶基因[37]。在TGF-β1诱导的EMT 体外模型,抑制ELK3表达可使EGR1和波形蛋白表达下调;抑制P38 MAPK磷酸化水平可抑制ELK3及EGR1的表达水平,甚至可能逆转EMT[36]。因此,MAPK/ELK3/EGR1信号途径可能参与肝纤维化EMT过程(图1)。
EGR1参与肝纤维化的机制具有模型依赖性。各种肝纤维化EGR1敲除小鼠模型肝组织损伤的表现可能不同。α-萘酯(α-naphthylisothiocyanate)饮食模型敲除EGR1,肝炎症反应及肝损伤无显著变化,但Ⅰ型胶原蛋白表达水平显著增加[38];CCl4模型肝组织的肿瘤坏死因子α(tumor necrosis factorα,TNF-α)mRNA和蛋白表达水平降低,胶原蛋白沉积水平升高,且肝损伤加重[37-39];对乙酰氨基酚模型肝组织的胶原蛋白表达和分布水平增加[33]。与以上模型相反,胆管结扎(bile duct ligation,BDL)模型肝组织的胶原蛋白沉积未受影响,炎症反应和肝损伤反而减轻[40]。
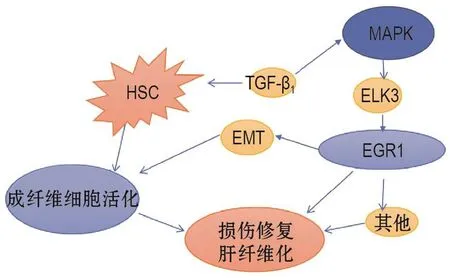
图1 早期生长反应蛋白1(EGR1)在肝纤维化的肝星状细胞活化通路.MAPK:丝裂原活化蛋白激酶;ELK3:ETS转录因子;TGF-β1:转化生长因子β1;EMT:上皮-间充质转化;HSC:肝星状细胞.
这些不同表现的原因可能是EGR1在不同模型的作用机制不同。如在α-萘酯模型,EGR1缺乏可抑制促纤维化基因整合素β6 mRNA的表达,该基因可结合并激活TGF-β1,故EGR1敲除在一定程度上抑制纤维化发展[38]。而CCl4模型可能与TNF-α表达水平减少或肝伤口愈合失调有关,这削弱了肝的保护机制[37]。
3.4 胆汁淤积性肝病
胆汁淤积是指在肝细胞或胆管水平发生的胆汁生成、分泌或流动障碍,引起细胞内胆汁酸积聚,造成肝细胞损伤。若持续发生肝细胞损伤,可发展成胆汁纤维化或肝硬变[40]。胆汁酸大量积累引起肝细胞炎症细胞因子表达水平增强与胆汁酸激活EGR1有关[41]。目前报道,胆汁酸经MAPK下游信号通路,激活EGR1,促进促炎介质释放,肝细胞炎症细胞因子表达水平提高,导致肝细胞损伤[42]。另外,在胆汁淤积性肝病患者[43]、BDL模型[40]的肝组织以及小鼠原代肝细胞[38]均发现EGR1水平升高。因此推测,EGR1与胆汁淤积的肝纤维化可能具有重要联系。
SHP由于缺乏保守的DNA结合结构域,不能直接进行转录的调控。但活化的SHP可在特定组织抑制转录因子的活性。SHP对参与肝代谢过程的基因具有负调控作用[44],对胆汁淤积的肝纤维化还有预防作用[45]。EGR1和SHP基因启动子均可被HNF4α直接激活;活化的SHP可抑制EGR1启动子的活性及其基因的转录水平[46]。虽然BDL小鼠胆汁淤积肝纤维化模型和人肝硬变组织中HNF4α和EGR1表达增加,SHP表达降低,但HNF4α敲除小鼠肝组织EGR1 mRNA和蛋白水平仍升高。由此推测,EGR1表达增加的原因,除SHP表达降低减少对EGR1抑制及HNF4α表达升高增加对EGR1激活外,可能还有其他分子激活了EGR1。
肝再生增强剂(augmenter of liver regeneration)是一种抗凋亡因子,既可减少游离脂肪酸引起的脂肪细胞凋亡[47],也可改善胆管结扎后大鼠的线粒体和肝功能。EGR1可与肝再生增强剂启动子的反应元件特异性结合,增加肝再生增强剂的表达。但在胆汁淤积的肝病中,EGR1被胆汁酸激活的同时削弱了与肝再生增强剂启动子结合能力,使肝再生增强剂启动子活性降低,可能导致胆汁淤积的肝功能恢复受阻[48]。
E2F1可促进肝细胞的糖酵解和脂质生成[49],并且E2F1基因敲除小鼠胆汁淤积肝纤维化减少。而在胆汁淤积过程E2F1直接激活EGR1转录,但这一过程可被SHP抑制[3]。综上所述,EGR1在胆汁淤积性肝组织被HNF4α,SHP或E2F1等激活表达,可能进一步引起肝细胞损伤或肝纤维化(图2)。
3.5 酒精性肝病
酒精性肝病(alcoholic liver disease,ALD)可发展为脂肪性肝炎、肝纤维化或肝硬变,导致肝功能衰竭或死亡的危险性增加。
3.5.1 酒精诱导的肝脂肪变性
EGR1可能是酒精引起ALD发生脂肪变性的主要因素[17]。EGR1可与脂肪变性相关基因启动子结合,如 TNF-α[50]、甾醇调节元件结合蛋白 1c(sterol regulatory element binding protein-1,SREBP-1c)[50-51]。例如,EGR1诱导TNF-α的高表达可促使SREBP-1c成熟,促进肝脂质的合成[52]。EGR1敲除小鼠仅部分阻断酒精引起的甘油三酯积累,并不能阻止肝脂肪变性,而EGR1和SREBP-1c双重敲除小鼠可消除肝脂肪变性[50]。这些研究表明,EGR1不是影响SREBP-1c表达的唯一分子通路(图2)。
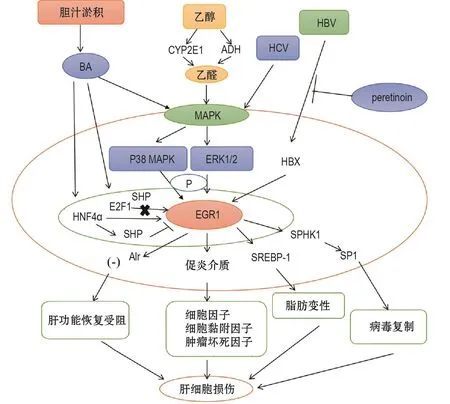
图2 早期生长反应蛋白1在胆汁淤积性肝病、酒精肝及病毒性肝炎的上下游通路模式.BA:胆汁酸;CYP2E1:细胞色素P450 2E1;ADH:醇脱氢酶;HCV:丙型肝炎病毒;HBV:乙型肝炎病毒;MAPK:丝裂原活化蛋白激酶;ERK1/2:细胞外信号调节蛋白激酶1/2;HBX:乙型肝炎病毒X;P:磷酸化;E2F1:E2F1转录因子;SHP:小异二聚体伴侣;HNF4α:肝细胞核因子4α;Alr:肝再生增强剂;SREBP-1:甾醇调节元件结合蛋白1;SPHK1:鞘氨醇激酶1;SP1:鞘氨醇-1-磷酸.
蛋白酶体(proteasome)是一种大分子复合物,可选择性降解细胞内的蛋白质[53],其活性降低可提高EGR1在细胞内的稳定性,可能引起酒精诱导肝组织EGR1表达水平升高[54]。但有研究发现,在酒精诱导的急性肝损伤肝组织,EGR1表达水平升高,蛋白酶体活性却无明显变化。因此,EGR1表达水平增加可能不是由蛋白酶体活性下降引起的[55]。多数研究认为,酒精氧化生成的乙醛可能是激活EGR1启动子、增强EGR1基因转录和蛋白表达水平的主要原因[55-56]。
3.5.2 酒精诱导的慢性或急性肝损伤
在酒精刺激下,脂多糖刺激巨噬细胞(如库普弗细胞)产生各种炎症因子(如TNF-α和白细胞介素1等),加剧肝损伤[57]。另外,脂多糖刺激引起ERK1/2磷酸化,有助于提高EGR1在库普弗细胞的表达水平,并增加产生TNF-α[58]。在4周的慢性酒精脂肪肝病模型,EGR1敲除小鼠既未表现出肝脂肪变性,也未出现TNF-α表达水平升高[51]。因此推测,EGR1敲除可能对慢性酒精性肝损伤具有预防作用。然而,在造模18个月的LE(Long-Evans)大鼠肝组织中EGR1表达水平显著提高;EGR1还促进促纤维化基因表达以及增强SREBP-1c蛋白的转录活性[59]。因此,慢性酒精性肝损伤模型经长期酒精诱导,EGR1表达水平可能升高,并通过与SREBP-1c启动子结合,促进肝组织脂肪变性(图2)。
此外,在急性酒精性肝病EGR1敲除小鼠与野生型小鼠中,前者肝组织中甘油三酯水平低30%,脂质过氧化物水平以及血清的酒精含量二者相当,但前者谷丙转氨酶水平更高且肝损伤较严重[55]。因此,EGR1敲除在急性酒精性肝损伤可能不影响酒精代谢但可能部分阻止脂肪变性。
综上所述,EGR1表达可能促进脂肪变性甚至纤维化的发展,但在急性酒精性肝病,缺乏EGR1可能会加剧肝损伤。
3.6 病毒性肝炎(乙型肝炎和丙型肝炎)
EGR1与病毒性肝炎的相关研究主要集中在乙型肝炎病毒(hepatitis B virus,HBV)和丙型肝炎病毒(hepatitis C virus,HCV)。一种非环状维A酸(peretinoin)可抑制HBV或HCV的复制[60-61]。EGR1在HBV复制的Huh7细胞被诱导,但peretinoin可阻断EGR1下游通路,如EGR1-鞘氨醇激酶1(sphingosine kinase 1,SPHK1)-鞘氨醇-1-磷酸(sphingosine-1-phosphate,SP1)信号通路,最终抑制HBV复制[60]。此外,HCV无包膜颗粒的内吞作用可激活ERK1/2,磷酸化ERK1/2激活EGR1,可能参与HCV感染细胞的过程[62]。EGR1上调可促进鼠肝炎病毒进入小鼠星形细胞瘤的细胞,并提高感染的持续性[63]。综上可见,下调EGR1也许有助于抑制肝炎病毒的复制。
3.7 肝癌
肝癌(肝细胞癌,hepatocellular carcinoma,HCC)是肝最常见的恶性肿瘤,也是第三大致死性癌症。EGR1与HCC相关机制的报道存有分歧,如有些发现EGR1在HCC中过表达,有些则发现表达下调[64]。
3.7.1 EGR1可作为抑癌基因
EGR1可抑制HHCC细胞生长速度,减少S期细胞,降低其致瘤性。但EGR1未抑制SMMC7721细胞的恶性表型,其机制尚不清楚[65]。另外,YANG等[64]运用[125I]UdR放射性核素联合干扰素γ的基因治疗手段,激活EGR1启动子活性可有效减少肿瘤的增殖,提高H22细胞存活率。EGR1抑制肿瘤的机制主要通过激活肿瘤抑制因子,包括P53、P73、纤连蛋白和PTEN等(图3)。ZHAO等[66]发现,沉默EGR1,3′-叠氮基-3′-脱氧胸苷(3′-azido-3′-deoxythymidine)联合 As2O3治疗HCC 效果减弱,癌细胞凋亡标志物(如P53)表达减少。
3.7.2 EGR1可作为癌基因
激活EGR1可能增加HCC细胞生长、侵袭和迁移的能力[67]或促进HCC细胞自噬作用,导致HCC细胞耐药性增强[68](图3)。如红外线刺激EGR1表达的快速上调,可降低HCC细胞对红外线治疗的敏感性,促进HCC细胞迁移[69]。自噬可清除受损的细胞器,抑制癌症发生;但在辐射、化疗和氧化应激等应激下,自噬也可促进癌细胞的存活[70]。如在低氧条件下,EGR1可与微管相关蛋白1轻链3的启动子结合,促进HCC细胞自噬体形成[68];或通过促进低氧诱导的自噬来降低肿瘤细胞对抗癌药物的化学敏感性,增强HCC细胞的耐药能力[71]。故推测抑制EGR1的表达水平可能会提高抗癌药物在低氧下的药效作用。EGR1在肝细胞生长因子(hepatocyte growth factor,HGF)诱导的HCC细胞侵袭时过表达,并激活HGF/c-Met的下游靶点基质金属肽酶,提高HCC细胞侵袭能力[72]。
3.7.3 EGR1与病毒性肝炎相关HCC
目前研究主要集中在HBV或HCV相关HCC。乙型肝炎病毒X(hepatitis B virus X,HBX)蛋白的表达可提高病毒启动子和增强子的活性,促进HBV的转录和复制过程[73]。早期抑制HBV感染肝细胞的炎症介质(如前列腺素E2)产生,可减轻肝细胞恶性病变和减少肝癌的发生[74]。HBX可促进EGR1与微粒体前列腺素E合酶1启动子区域特定位点结合,激活微粒体前列腺素E合酶1启动子的转录活性,增加HBV感染肝细胞的炎症介质产生,促进HCC 的发生。ZHANG等[75]认为,HBX 在NF-κB/EGR1通路中上调EGR1,并通过促进miR-3928v(一种小分子RNA)的表达来抑制肿瘤抑制基因的表达,加速HCC的发展。另外研究发现,HCV核心蛋白可激活胰岛素样生长因子Ⅱ(insulinlike growth factor Ⅱ,IGF-Ⅱ);IGF-Ⅱ的过表达可刺激HCV感染的肝癌细胞的生长[76]。故EGR1作为IGF-Ⅱ基因转录调节的直接靶标,可能参与HCV相关HCC的发生和发展。
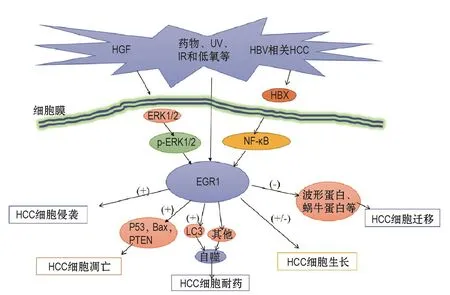
图3 早期生长反应蛋白1在肝细胞癌的上下通路模式.HGF:肝细胞生长因子;UV:紫外线;IR:红外线;p-ERK1/2:磷酸化的细胞外信号调节蛋白激酶1/2;LC3:微管相关蛋白1轻链3;PTEN:第10号染色体缺失的磷酸酶和张力蛋白同源物基因;HCC:肝细胞癌.
综上所述,也许基于HCC发生存在复杂的异质性,EGR1既可能表现抑癌作用,也可能表现促癌作用。EGR1在HCC的表达水平可能可以作为临床HCC复发的生物学标志分子。另外,尝试通过抑制EGR1表达来增加肿瘤细胞对化疗药物敏感性可能可提供治疗HCC的新方法。
4 结语
一方面,MAPK,SHP,E2F1,HNF4α及TGF-β1等多种上游分子可调控EGR1的表达;另一方面,EGR1可与下游不同靶基因的启动子(如与细胞凋亡或纤维化发生等相关基因)特异性结合,诱导靶基因转录表达。通过上下游分子相互作用,EGR1不仅参与肝再生、细胞生长分化以及凋亡等作用,也参与肝纤维化、酒精性肝病、肝癌以及病毒性肝炎等的病变。在不同的生理、病理条件下,EGR1既可能参与阻止肝组织病理改变,也可能加速肝病发展,其原因尚不清楚,推测可能与EGR1具有负反馈调节机制或EGR1调控各类下游靶基因及促进细胞生长和凋亡的生物学功能有关。目前,EGR1研究主要集中在非临床模型,如继续在临床研究中观察其调控机制,将有助于进一步阐明EGR1对肝病治疗的重要意义。
猜你喜欢
传染病信息(2022年3期)2022-07-15 08:24:28
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
肝博士(2021年1期)2021-03-29 02:32:16
学苑创造·A版(2020年12期)2020-01-07 14:07:23
中国外汇(2019年15期)2019-10-14 01:00:34
作文教学研究(2016年1期)2016-07-05 12:22:47
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
哈尔滨医药(2015年4期)2015-12-01 03:57:54
医学研究杂志(2015年6期)2015-07-01 17:40:08
医学研究杂志(2015年8期)2015-06-22 14:00:57
