
Enhancement in antinociceptive and antiinflammatory effects of tramadol by transdermal proniosome gel
2021-01-19 04:56HirlShhSheryJobTmerShehteMohmeAlyMorsyf
,Hirl Shh,Shery Job,Tmer M.Shehte,Mohme Aly Morsyf
aInstitute of Pharmacy,Nirma University,Ahmedabad 382481,India
b College of Clinical Pharmacy,King Faisal University,Al-Ahsa 31982,Saudi Arabia
c Arihant School of Pharmacy&BRI,Gandhinagar 382421,India
dCollege of Pharmacy,Gulf Medical University,Ajman 4184,United Arab Emirates
eFaculty of Pharmacy,University of Zagazig,Zagazig 44519,Egypt
f Faculty of Medicine,Minia University,El-Minia 61511,Egypt
Keywords:Tramadol Proniosomes Flux Edema Pharmacokinetics Rats
ABSTRACT Oral therapy of tramadol,an opiate analgesic,undergoes extensive hepatic metabolism and requires frequent administration.Transdermal therapy by virtue can overcome these issues and can improve the efficacy and reduce abuse liability of tramadol.The aim of this research was to investigate the possibility of transdermal delivery of tramadol by formulating proniosome gel and evaluate its therapeutic potential in vivo.The effect of formulation composition as well as amount of drug on physicochemical characteristics of prepared proniosomes were examined.Best proniosome gel(F4)was selected and evaluated for drug release,stability and transdermal efficacy by ex vivo and in vivo experiments.The vesicles demonstrated optimal properties including spherical shape,nanosize with good entrapment efficiency,adequate zeta potential,higher stability and greater transdermal flux.The amorphization and dispersion of tramadol in the aqueous core of proniosome vesicles was confirmed by differential scanning calorimeter.Release profile of F4 was distinct (P <0.001) from control and displayed steady and prolonged tramadol release by Fickian diffusion.Transdermal therapy of F4 showed prominent reduction of induced twitches (P <0.005) in mice and edema (P <0.05) in rats,as compared to oral tramadol.The improvement in clinical efficacy of tramadol in transdermal therapy is correlated with the pharmacokinetic data observed.In conclusion,the observed improvement in antinociceptive and anti-inflammatory effects from proniosome carriers signifies its potential to be a suitable alternative to oral therapy of tramadol with greater efficacy.
1.Introduction
Tramadol is a centrally acting,synthetic,opiate analgesic that relieves pain by binding to the μ-opioid receptors and by inhibiting the reuptake of two pain modifying neurotransmitters,serotonin and norepinephrine [1].Tramadol is indicated in acute and chronic pain of moderate to severe intensity in patients of all ages [2].This drug is available as oral formulations and injections.In oral therapy,tramadol is administered very frequently at every 4 to 6 h as the terminal elimination half-life (T½β) is approximately 6 h[3].However,orally administered tramadol generally elicits opioid-like effects (both mentally and physically),although these responses are mild and not observed in parenteral therapy [4,5].Furthermore,oral administration of tramadol primarily undergoes significant hepatic first-pass metabolism,with an approximately 10% to 30% is excreted unchanged in urine.On the other hand,parenteral administration is associated with number of inherent complications including patient discomfort,phlebitis,thrombosis and fluctuation in plasma drug concentration after multiple injections [6].In this context,an alternative route for tramadol delivery is most appropriate.
Non-invasive transdermal route of administration proposes unique advantages such as ease of application,improved potency and patient compliance by providing a steady state plasma drug concentration for extended period [7].Various transdermal systems and enhancement approaches are used to achieve effective drug delivery through the skin [8].In case of tramadol,the transdermal delivery system can evade probable drug dependence and abuse risk potential by preventing peak and trough plasma concentrations and decreasing drug accumulation [9,10].In addition,this delivery route can overcome the bitter taste and also overcome the first pass effect of tramadol.Nevertheless,the choice of transdermal system depends on drug’s physicochemical properties as well as its therapeutic use [11].The properties of tramadol including low molecular weight (299.8 Da),adequate aqueous solubility (>1 mg/ml),good partition coefficient (logP 1.35),low melting point(180 °C),and short half-life (6 h) seems ideal for transdermal therapy.However,requirement of high daily dose(50 mg)and relatively close therapeutic blood levels (0.1-0.3 mg/l) and toxic level between 1 and 2 mg/l of tramadol requires specific transdermal delivery systems.Literature suggests that couple of studies have been carried out to deliver tramadol through skin.In one approach,a matrix transdermal film of tramadol was developed and evaluated for its potential to provide steady drug concentration[10].In another attempt,a physical enhancement approach (iontophoresis) has been utilized to enhance drug flux in order to achieve required therapeutic level[12].Therefore,a suitable transdermal system which can transport greater amount of tramadol in a controlled fashion is ideal for its effective delivery.
Nanocarriers have demonstrated their potential to improve the percutaneous absorption of various drug molecules [13,14].Surfactant based colloidal drug carriers such as niosomes and its hybrid (proniosomes) are excellent tools for transporting greater amount of drug across skin and can release at controlled rate for systemic absorption [15].Dry proniosomes formulations are transformed to niosomes after hydration which in turn serves as an innovative drug delivery system for percutaneous drug administration [16].In addition,proniosomes exhibits excellent stability owing to its potential to surmount physical instability including aggregation or fusion,leaking of entrapped drugs,and sedimentation.In topical delivery,niosomes can augment the drug permeation by overcoming the barrier property and can form drug depot[17].Proniosomes are generally incorporated in suitable gel where it hydrates to form niosome gel for transdermal application.To our knowledge there is no investigation so far evaluated the potential of proniosomes to deliver tramadol through transdermal route.Accordingly,the objective of present study was to develop tramadol loaded proniosome gel and evaluate its feasibility for transdermal therapy.Effect of various formulation ingredients as well as drug content on physicochemical characteristics of prepared proniosomes were assessed.The selected proniosome gel(F4)was characterized,evaluated for drug release,permeation and stability.In vivostudies were performed for evaluating antinociceptive and anti-inflammatory effect as well as pharmacokinetic parameters using F4 and compared with marketed oral tablet.
2.Materials and methods
2.1.Chemicals and reagents
Tramadol HCl was a gift sample from Zydus Cadila Ltd.(Ahmedabad,India).Soya lecithin and cholesterol were gifted by CP Kelco UK Ltd (Surrey,UK).Span20,Span40,Span60,Span80,Tween20,Tween40,Tween60,Tween80 and hydroxypropyl methylcellulose (HPMC) were procured from Central Drug House Pvt.Ltd.(Mumbai,India).Dialysis membrane,membrane filter (0.22 μm),syringe filter (0.22 μm)were purchased from Merck India Ltd.(India).All other chemicals and reagents used in the development of formulations were analytical grade.
2.2.Analytical method
Quantification of tramadol in various samples was performed by high-performance liquid chromatography (HPLC)system (Shimadzu LC-20AT,Tokyo,Japan) consisting of chromatographic column with LiChrospher 60 RP-select(250 mm×4 mm,5 μm,Merck,Germany).Chromatographic separation was carried out by a mobile phase consists of acetonitrile-0.01 M phosphate buffer (3:7,v/v) at pH 2.8 [18].The injection volume was 100 μl and the elution was done isocratically at 25°C at a flow rate of 1 ml/min with fluorescence detection (λex264 nm/λem344 nm).The analytical method was validated for system suitability,sensitivity,selectivity,linearity,precision and accuracy.The validated method has a retention time of 4.2 min with a linearity in the range of 10-400 pmol/ml(r2=0.992).The limit of detection was found to be 5.9 pmol/ml and the limit of quantitation was 39 pmol/ml.Plasma samples were allowed to protein precipitation with same volume of acetonitrile and 2 propanol,centrifuged at 10 000 rpm (10 min) and the supernatant was filtered using 0.22 μm filter (syringe filter,Merck,India) and injected (100 μl) into the HPLC.The sample withdrawn at time zero served as the baseline value.The percentage recovery of tramadol in plasma (86%) was determined by spiking different concentrations of drug(100-10 000 ng) in plasma,thoroughly mixed by vortexing(2 min),and the tramadol content was determined similarly as described before.
2.3.Preparation of proniosomes
Proniosomes were formulated by coacervation phase separation technique with slight modification as reported earlier using various types and concentrations of non-ionic surfactants,lecithin and cholesterol [17].Suitable quantities of proniosome components and tramadol were mixed with absolute ethyl alcohol (2.5 ml) in a glass vial.After incorporating all components,the vial was covered to avoid evaporation of solvent and heated in a thermostatic water bath (65 ± 5°C) for 5 min to achieve complete solubility of surfactants.Further,1.5 ml phosphate buffer (pH 7.4)was added and the heating was continued on the water bath (~2 min) to get transparent solution.The solution was further cool down to room temperature so that the dispersion transformed to a clear proniosome liquid.Finally,the solutions was mixed with suitable quantity of HPMC(2%,w/w) to obtain 0.5% (w/w) tramadol concentration.Similarly,placebo proniosome gel were prepared using HPMC (2%,w/w) without the drug.The control formulation was prepared using HPMC (2%,w/w) containing tramadol(0.5%,w/w).
2.4.Evaluation of proniosomes
2.4.1.Entrapment efficiency
Prepared proniosomes (0.1 g) was hydrated with phosphate buffer saline (pH 7.4;10 ml) in a glass vial.The aqueous dispersion was stirred using ultrasonicator (Transonic T460/H,Elma,Germany) for 30 min.The obtained noisome components were isolated from free drug by a high-speed cooling centrifuge (Sigma 3K30 refrigerated centrifuge,Laborzentrifugen,Germany) moving at 15,000 rpm at 25 ± 0.1°C for 15 min.The supernatant fraction containing free tramadol was analysed by HPLC technique.The vesicular aggregate residue was washed thrice with phosphate buffer saline and assayed similarly.Separately,entrapped drug in vesicular aggregates was also determined.The percentage entrapment efficiency (EE%) was computed using following equation:
EE%=[(Ct-Cr)/Ct]×100
whereCtis total andCris free tramadol content.
2.4.2.Particle size and zeta potential
The particle size of vesicles was measured by light scattering based on laser diffraction utilizing Mastersizer (Model S,Ver.2.15;Malvern Instruments,Malvern,UK).Laser diffraction was checked at 25°C at a scattering angle of 90°.Size of proniosomes was determined from a total of 100 particles of 10 cycles and polydispersity index was recorded.The zeta potential of proniosomes was carried out with a Zetasizer(Malvern Zetasizer Nano ZS,Malvern,UK).It was measured for individual sample after dispersing in water for hydration and subsequent ultrasonication for 5 min[19].
2.4.3.Microscopy
Proniosomes (0.2 g) was hydrated with phosphate buffer(pH 7.4;10 ml) and few drops of niosome dispersion were placed on a glass slide and viewed under optical microscope.Structural features of proniosomes was investigated with transmission electron microscopy (TEM,Tecnai 20,Philips,Holland) operated at 200 kV.Proniosomes were hydrated and stained with phosphotungstic acid solution (2%,w/v).The samples were placed directly on copper grids,dried at room temperature(25±2°C)and TEM images were captured.
2.4.4.Differential scanning calorimetry(DSC)
The thermal characteristics of tramadol HCl,physical mixture,placebo proniosomes and drug entrapped proniosomes were determined by DSC (DSC 60,Shimadzu,Japan).Samples were placed in aluminum crimped pans and sealed nonhermetically,and thermograms were recorded over the temperature of 50-300°C at a heating rate of 10°C/min.
2.5.In vitro release
The drug release studies were performed employing a vertical Franz diffusion cell having an effective surface area of 1.13 cm2and phosphate buffer saline (10 ml) as receptor medium.Briefly,weighed quantity of proniosome gel (F4)equivalent to 50 mg of tramadol HCl was taken on a previously soaked cellophane dialyzing membrane (MWCO 12-14 kDa,Spectra/por® Spectrum Laboratories Inc.,Berkeley,CA) that separates the donor and receptor cell.The entire assembly was placed on a thermostatically controlled water bath set at 37 ± 0.1°C and receptor medium was stirred at 50 rpm[20].Aliquots of sample (1 ml) were drawn at regular time intervals and replaced with same volume of fresh media.The samples were subsequently diluted and analyzed for tramadol content by HPLC.Similarly,control experiments were run in parallel for tramadol gel prepared using HPMC.The data were analyzed to determine correlation coefficient (r2) and release kinetics in all cases[21].
2.6.Skin permeation
Intact full thickness skin of New Zealand Wistar albino rats was prepared by removing the fat and subcutaneous tissues by applying isopropyl alcohol and using scalpel.The skin was cleaned with water and stored at-20°C until used [22].A similar experimental setup used in release studies was employed in skin permeation experiments.Skin was mounted on diffusion cell with the stratum corneum facing up.The amount of tramadol permeated via the skin membrane into receiver media was determined using HPLC.Steady state flux,Jss(μg/cm2/h),was estimated from the slope of linear plot between cumulative amount permeated and time[23].
2.7.The rate of spontaneity
Instantaneous formation of niosomes was evaluated by hydrating proniosome gel (250 mg) with phosphate buffer saline (5 ml) taken in a glass vial.A drop of dispersion was transferred with an Eppendorf pipette and placed on the Neubauer chamber (Blaubrand,Wertheim,Germany).The number of niosome vesicles visible in square millimeter area was counted under a research microscope (Nikon Eclipse LV 100 Pol)as elaborated by Thakur et al.[24].
2.8.Stability
Physical and chemical stability of F4 were determined after transferring in glass vial,sealed and placed in a desiccator.The products were stored for a period of 3 months at controlled room temperature (25 ± 1°C),refrigeration temperature (4-8°C) and maintained a relative humidity of 75 ± 5% using saturated solution of sodium chloride.Vesicle size,EE% and percentage drug retention of samples was analyzed at 0,1,2 and 3 months after storage.The niosome formulations were evaluated optically for physical stability as well as for both free and entrapped drug[25].
2.9.Anti-inflammatory effect
Animal experiments were done as per the standards and guidelines set by the institutional animal ethical committee(Protocol No.ASP&BRI/AH/2013/07) at the Arihant School of Pharmacy&Bio-research Institute,Gujarat,India.All animals were fedad libitumand housed in light and dark cycle in an ambient temperature-controlled environment.Acute type of paw edema was induced in New Zealand Wistar albino rats (200 ± 50 g each) via subcutaneous injection (0.1 ml) of formaldehyde (4%) at the left sole of the rat’s hind paw half an hour prior to drug dosing.The animals were divided into four groups,each comprised of 6 rats.
Group I:Negative control
Group II:Positive control(Marketed tablet of tramadol HCl as standard)
Group III:Placebo(Tested proniosome gel without tramadol HCl)
Group IV:Treated group (Tested proniosome gel with tramadol HCl;F4)
For group II,tablet was dispersed in phosphate buffer saline (2 ml) and administered orally to rats via intra-gastric gavage.The dose of tramadol HCl administered (7.14 mg)was calculated according to the equation described in the literature [26].For groups III and IV,dorsal skin region of rats was shaved and gel were applied uniformly on the skin using a custom-made applicator.Group III was applied with placebo gel,while group IV received proniosome gel(F4,weight equivalent to 7.14 mg of tramadol HCl).An open-ended circular holder (diameter 2 cm) was positioned above applied area to enclose the gel and was covered with Parafilm.The anti-inflammatory response of all groups was determined by measuring paw edema volume using digital micrometer gauge (Mitutoyo,Japan) at specific time intervals.Furthermore,for group II and group IV,blood samples were withdrawn (~200 μl) from the tail vein using dry heparinized tubes at 1,2,4,6,8,12 and 24 h.Samples were allowed to protein precipitation with same volume of acetonitrile and 2-propanol and tramadol content was determined using HPLC.The pharmacokinetic parameters were calculated using non-compartmental pharmacokinetic model.Pharmacokinetic variables like area under the concentration-time curve (AUC),peak concentration(Cmax),and time to peak concentration(Tmax)were determined.
2.10.Antinociceptive activity
In antinociceptive activity test,mice(25±5 g)were randomly separated into three groups and individual group consists of six animals.The animals were categorized as;group I:negative control,group II:positive control (marketed tablet of tramadol HCl as standard) and group III:treated group(tested proniosome gel with tramadol HCl;F4).A dose of 4 mg was administered in a similar method as described in antiinflammatory effect section.Analgesic effect was assessed on acetic acid-induced abdominal constriction.Aqueous solution of acetic acid(0.7%,v/v;10 ml/kg)was injected through intraperitoneal route 30 min after applying the medicated topical formulation [27].All mouse was kept in separate transparent inspection cage.Abdominal muscle constriction that takes place between 5 and 15 min after acetic acid injection was recorded.Activity was considered as percent inhibition of muscle twitching among negative control,positive control and tested groups [28].Pharmacokinetic data was assessed for group II and group III.Blood samples were withdrawn(~100 μl)from the tail vein using dry heparinized tubes at 30,45,60,and 75 min after dose administration.Samples were allowed to protein precipitation with same volume of acetonitrile and 2-propanol and tramadol content were assayed using HPLC.
2.11.Skin irritation
Skin irritancy study was conducted by applying a single dose (100 mg) of F4 (2.56 mg tramadol HCl) to the shaved dorsal side of male albino rabbits (1.75 ± 0.25 kg).The other side was further subdivided into two areas,one applied with standard skin irritant (2.5% sodium dodecyl sulphate)which was considered as a positive control and one received placebo proniosomes (negative control).The tested area was monitored and graded for erythema and edema for a period of 7 days.The test score was assigned according to Draize dermal scoring criteria[29].
2.12.Data analysis
The statistical evaluation of the data was analyzed using one-way ANOVA and Turkey’s multiple comparison posttest (SPSS 23,SPSS Inc.,Chicago,IL,USA).The statistical differences between values showingP <0.05 were considered as significant.
3.Results and discussion
Proniosomes are formed,when a certain ratio of three different phases such as surfactant,alcohol,and water are mixed.This proniosomes can be spontaneouslytransformed to stable niosome dispersion after dilution with aqueous phase.Typically,the proniosome gel formation occurs,when the hydrophilic groups of the surfactants interact with water and the lipophilic chains transform into lyotropic liquid crystals [30].In this study,proniosomes were successfully formulated by coacervation phase separation method using non-ionic surfactants,lecithin and cholesterol.The composition of various proniosome formulations prepared (F1-F10) are summarized in Table 1.Preliminary studies were performed to estimate the required quantity of various surfactants,lecithin,and cholesterol to prepare proniosomes.The effect of formulation ingredients (surfactants,lecithin and cholesterol) as well as amount of drug on physicochemical characteristics of prepared proniosomes were examined.

Table 1-Composition of prepared proniosomes.∗
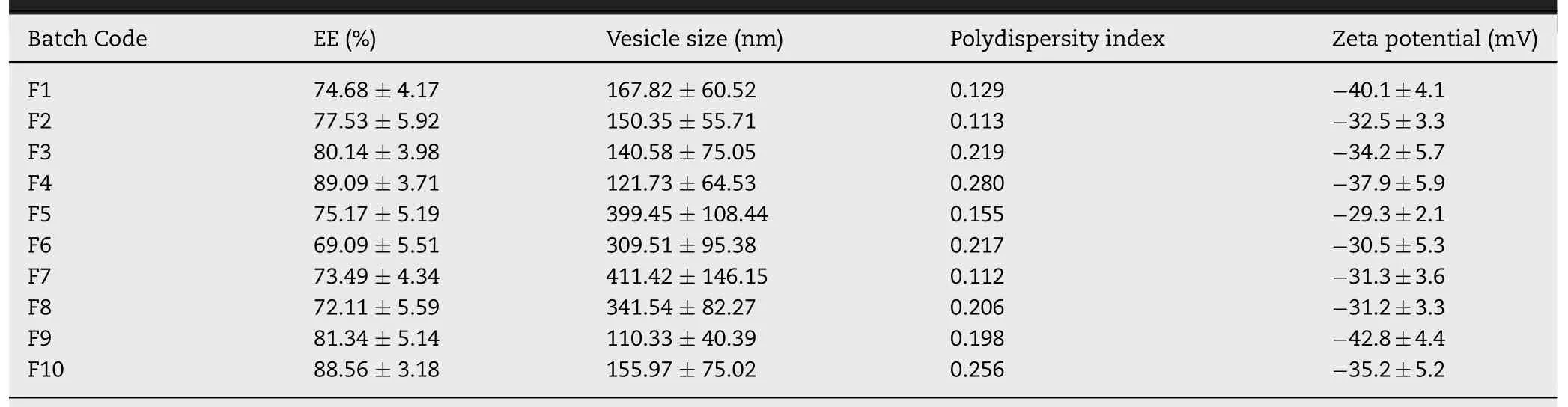
Table 2-Effect of formulation parameters on physicochemical characteristics of prepared proniosomes.
3.1.Entrapment efficiency
Vesicular carriers such as proniosomes and niosomes are comprised of non-ionic surfactants.The stability and EE%of niosomes vesicle were largely dictated by the inherent physicochemical characteristics of surfactant,such as HLB value,critical packing parameter,and phase transition temperature[31].The influence of using various conventional non-ionic surfactants on the EE%of proniosome preparations after hydration is displayed in Table 2.In case of niosomes prepared using Spans (F1,F2,F3,and F4),the EE% increases as;Span80>Span60>Span40>Span20.It is apparent from Table 2 that proniosomes prepared using Span80 showed significantly higher EE% (89.09%) than proniosomes prepared using other Span types (P <0.05).This is probably because the Span80 is a hydrophobic surfactant with low HLB value when compared with its counterparts[32].On the other hand,results from Table 2 also signifies that the EE%of proniosomes prepared using Tween series were comparable (~70%-75%),but significantly lower than their Span counterparts.Therefore,the above data demonstrate that decrease in HLB value of surfactant is likely to increases the EE%,under current experimental conditions.The observed difference in EE% between prepared proniosome formulations is probably due to the dissimilarity of chemical structure existing in their surfactants and their critical packing parameters which can eventually affect the bilayer structure of vesicles formed.Despite the fact that Span60 and Span80 possess approximately identical molecular weight (~430 g/mol),Span60 has a linear chain and higher HLB value (4.7) in comparison to Span80 (HLB:4.3) which might have reduced the encapsulation capacity of the drug.
In next stage,the effect of different tramadol content on EE% was assessed using Span80 as surfactant.It is evident from the Table 2 that increasing tramadol content from 60 mg(F9)to 100 mg(F4)in prepared proniosomes enhances the EE%considerably (81.34% and 89.09% for F9 and F4,respectively).Nevertheless,further increase in drug content (from 100 to 140 mg) did not significantly affect the EE% (Table 2).It was reported that fixed amount of vesicular components yield niosomes having specific entrapment efficiency besides precipitation of surplus drug[32].
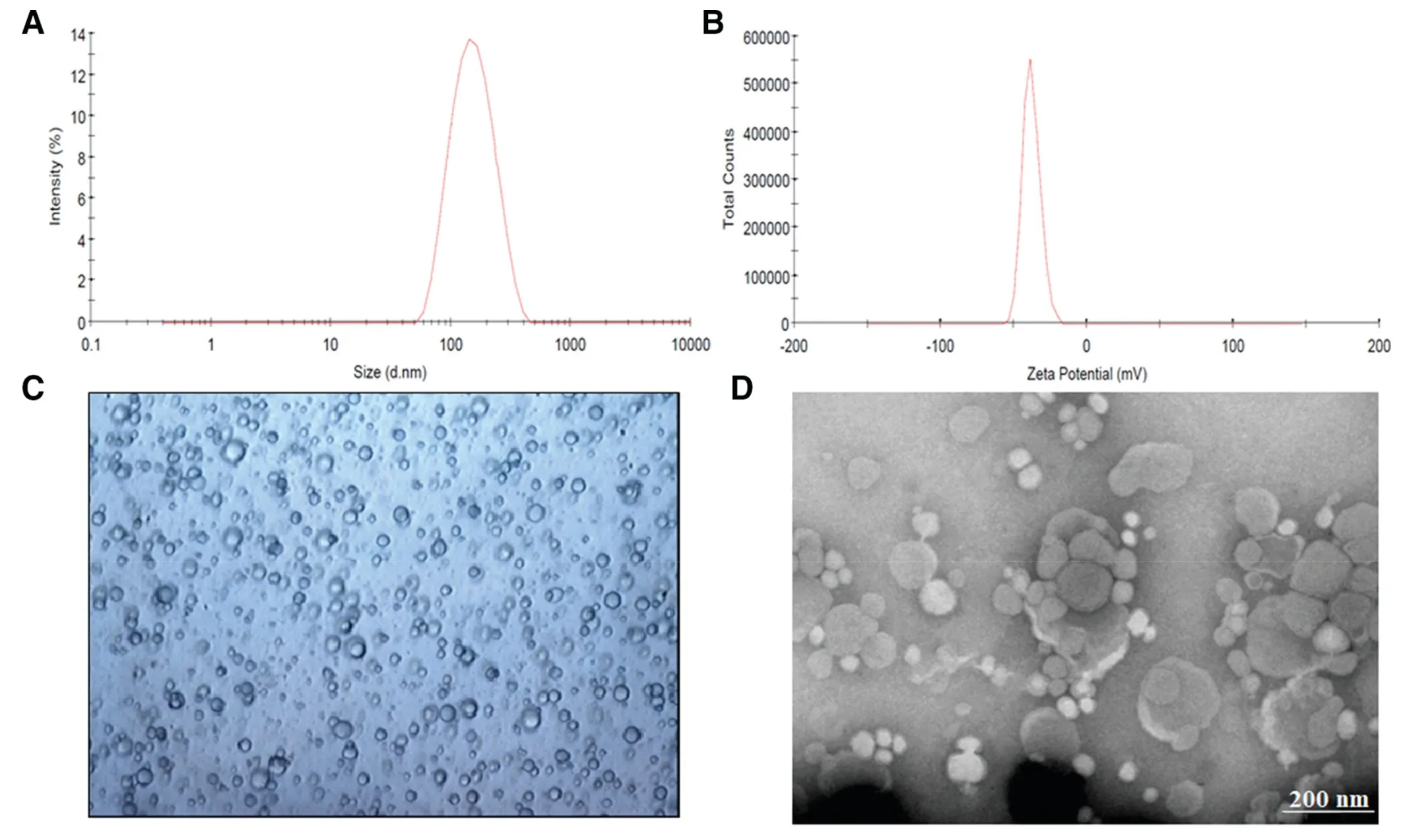
Fig.1-Characteristics of prepared tramadol proniosomes(formulation F4).(A)representative size distribution curve,(B)zeta potential distribution,(C)light microscopy picture(100×),and(D)transmission electron microscopy image.
3.2.Vesicle size and zeta potential
The mean vesicle diameter of prepared formulations(F1-F10)are summarized in Table 2.It is evident from the Table 2 that variation in vesicle size (110-170 nm) between proniosomes formulated with Spans were not significant.Particle size distribution curve of proniosomes (F4) was shown in Fig.1A,which demonstrates average vesicles size of 121 nm and narrow distribution of niosome particles.In contrast,niosomes prepared using Tweens (particle size 300-420 nm)were larger than those developed using Spans (P <0.05).The relationship observed between niosome size and Span hydrophobicity has been primarily attributed to the decrease in surface energy with increasing hydrophobicity,which eventually results in formation of smaller vesicles [33].This fact further clarifies the large vesicles formed in Tween based niosomes,which has comparatively lower hydrophobicity in comparison to Spans.
The effect of varying drug content on vesicle size was assessed from formulations F9 (60 mg),F4 (100 mg) and F10(140 mg).It seems there is a moderate relationship exists between the drug content and particle size.An increase in drug content from 60 to 140 mg increases the average particle size from 110 to 155 nm.This enlargement of vesicles size with increasing drug load is probably due to relatively high entrapment of tramadol HCl (polar drug) which in turn increase the aqueous content of the vesicle available for entrapping a large volume of drug solution as observed in earlier studies[34].
Zeta potential is significant since it determines the physical stability of colloidal vesicular carriers including proniosomes.The electrokinetic potential of prepared proniosomes ranged from-29 mV to-42 mV (Table 2).The data signifies that the zeta potential values of proniosomes prepared using Tweens (F5-F8) were relatively low (-29 mV to-31 mV) as compared to the values (-32 mV to-42 mV) of proniosomes prepared using Spans (F1-F4,F9,F10) (Table 2).Thus it seems the zeta potential value of proniosomes formulations increased with the hydrophilicity of the surfactants increased,which is also in agreement with earlier studies[35].
Fig.1B shows the zeta potential distribution of proniosomes(F4),which was-37.9±2.6 mV.It was found that all tested formulations demonstrated negative zeta potential due to the inclusion of negatively charged phospholipids and cholesterol.According to classical electrical double layer theory,zeta potential value above ±30 mV demonstrates moderate repulsion between similarly charged particles,thereby decreasing flocculation or aggregation and potentially stabilizes the dispersion.Therefore,the observed values are sufficiently high to contribute satisfactory repulsion between the vesicles and electrostatic stabilization.
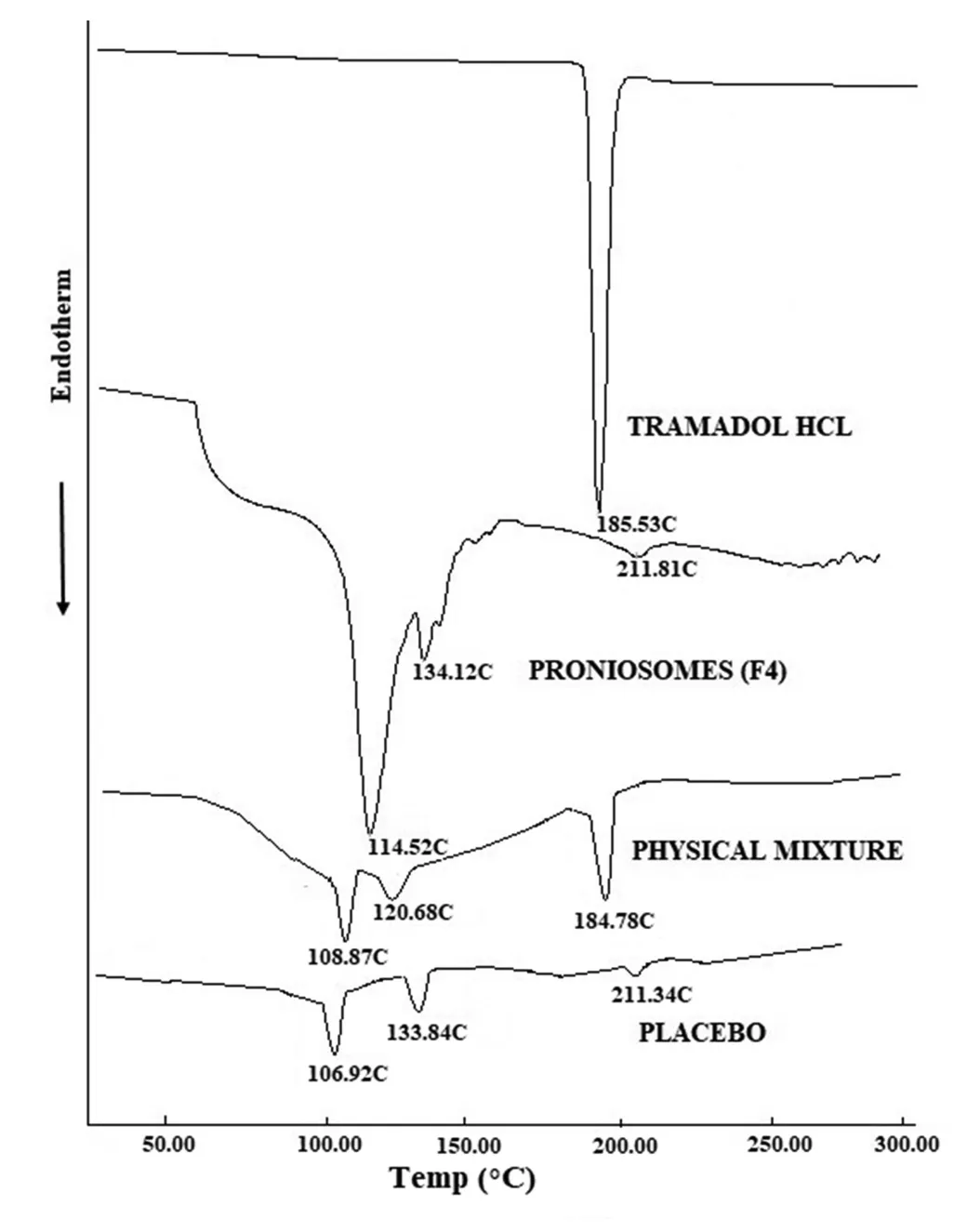
Fig.2-Differential scanning calorimetry patterns of tramadol,prepared proniosomes(F4),physical mixture and placebo formulation.
The polydispersity index value of 0-0.5 indicates homogenous,uniformly sized,spherical vesicles.As shown in Table 2,prepared formulations (F1-F10) have polydispersity index ranged from 0.112-0.280,which established that they were more uniform and homogenous.Moreover,the polydispersity index was not influenced by formulation composition studied.As proniosomes,F4,had shown greater EE%(89.09%±3.71%),it was chosen for additional evaluation,in vitrorelease,permeation,stability andin vivostudies.
3.3.Microscopy
Microscopic examination of the hydrated F4 displayed spherical,a vesicular structure with a central core of entrapped tramadol (Fig.1C).The TEM micrograph of F4 is illustrated in Fig.1D.It was noticed that the microscopic vesicles of niosomes formed after hydration of proniosomes are closed spherical bilayered structures,when viewed under TEM.
3.4.DSC
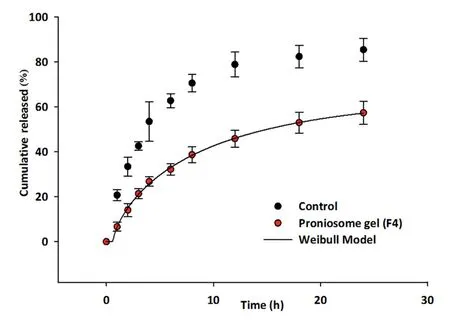
Fig.3-Comparison of percentage tramadol release from proniosome gel(F4)and control(tramadol gel prepared using hydroxypropyl methylcellulose).The data represents average±SD(n=6).
DSC thermogram of tramadol HCl,F4,physical mixture and placebo are presented in Fig.2.Tramadol HCl exhibited endothermic peak at 185.53°C indicating its melting point.The corresponding peak of drug is also observed in physical mixture thermogram at 184.78°C demonstrating the drugs’crystal structure.The absence of tramadol peak in F4 thermogram represent the evidence of tramadol amorphization and its dispersion in the aqueous core.Span80 in physical mixture showed an endothermic peak at 108.87°C,corresponding peak at 106.92°C and 114.52°C in thermogram of placebo and F4,respectively.The endotherm peak of cholesterol in F4 displayed at 134.12°C and at 133.84°C in placebo,was found to be shifted to 120.68°C in physical mixture signifying that all lipid components (cholesterol,lecithin and Span80)might interact each other while forming bilayer of the vesicles.The peak observed at 211.81°C in F4 and 211.34°C in placebo,may be attributed to the isotropic liquid phase of the lecithin as described by Yusuf et al.[36].
3.5.In vitro release
Thein vitrorelease studies was carried out for F4 that had highest drug EE%.Fig.3 compares the cumulative percentage of tramadol released at different time intervals from F4 and control gel.Two distinct release profiles were observed for F4 and control.In case of F4,the release was low and slow,but increased steadily over time (Fig.3).This observation is significant as an ideal proniosome gel should exhibit drug release for extended period so as to minimize repeated application.Literature indicates that major components of niosomes such as cholesterol can reduce drug leakage and lecithin can provide sustained drug release [37].In contrast,it is evident from Fig.3 that release of tramadol from control gel was biphasic with initial faster release followed by slower release and~60%of drug was released in 6 h(P <0.001).
Various mathematical models such as zero order,first order,Higuchi,Korsmeyer-Peppas,Weibull and Hixon-Crowell were used to evaluate the kinetics and mechanism of drug release from F4.Sum of square of residuals (SSR) values was 567.90,254.11,40.88,57.51,16.37 and 302.73 for Zero order,First order,Higuchi,Korsmeyer-Peppas,Weibull and Hixon-Crowell model,respectively.The release profile of F4 was fitted into Weibull model,which shown higher correlation coefficient(r2=0.9937,least SSR value(16.37)and F value (2.04) in comparison to Higuchi model.The equation for Weibull model isQ=1-exp [-(t)b/a]where Q represents quantity of drug released in time t,a represents the time constant and b represents the shape parameter.Therefore,it is concluded that release of tramadol from F4 depends on Weibull diffusion-controlled release pattern.In addition,diffusional exponent,nvalue<0.5 indicated Fickian release mechanism from F4.The results revealed that the dominant release mechanism is diffusion.

Fig.4-Comparison of amount of tramadol permeated at different time intervals across the rat skin membrane from proniosome gel(F4)and control(tramadol gel prepared using hydroxypropyl methylcellulose).The data represents average±SD(n=6).
3.6.Skin permeation
Theex vivopermeation investigation contributes invaluable information about formulation behaviour in thein vivoenvironment.Fig.4 compares the quantity of tramadol transported at specific time intervals across the rat skin membrane from F4 and control gel.It is obvious from the Fig.4 that the permeation rate of tramadol was significantly high (P < 0.001) from F4 in comparison to control.The steady state flux values of tramadol from F4 and control were 95.09 and 17.85 μg/cm2/h,respectively,signifies~5 fold enhancement in flux values with proniosomes.Various mechanisms are described in literature that can contribute in enhancing skin permeation of niosomes.Primary mechanism for improvement in transdermal drug permeation by topically applied surfactant vesicles is due to extraction of skin lipids or by disrupting the ordered structure of corneocytes upon binding to the keratin filament [38].Moreover,high thermodynamic activity of drug contributed by both adsorption and fusion of niosomes to the stratum corneum surface of the skin can facilitate drug permeation [39].Furthermore,the characteristics of prepared proniosomes such as low vesicle size,higher EE% as well as lipophilicity can cause these vesicles to efficiently fuse and permeate the skin.The low transition temperature of Span80 (due to intrinsic unsaturation of oleate) can also make it in the disordered state and completely fluid which in turn facilitate drug transport into and through the skin[33].
3.7.The rate of spontaneity
Spontaneity is referred as the numbers of niosomes generated quickly after proniosomes hydration.The rate of spontaneity of formulation F4 was~9.54×104.Smaller vesicle size (~121 nm) could explain the rapid hydration and subsequent rate of conversion of proniosomes to niosome vesicles.
3.8.Stability
The physical features,vesicle size,EE%,percentage drug retention was observed for F4 during storage at refrigerated(4-8°C) and room temperature (25°C) up to 3 months.At predetermined time intervals,the proniosomes gel was hydrated and noticed for the formation of spherical niosome vesicles with any indications of drug crystallization.Vesicle size,EE%and percentage drug retention values in the Table 3 establish that there was no significant alteration when stored at refrigerated temperature.However,it was observed that the formulation undergone physical and chemical instability at room temperature resulting in particle size enlargement,reduction of EE% and tramadol content due to drug leakage.It can be concluded from the stability studies that F4 was quite stable at refrigerated conditions than stored at room temperature.
3.9.Anti-inflammatory effect
Literature signifies that tramadol has known peripheral anti-inflammatory effect [40],accordingly this action was used to proof the efficiency of F4.At present,there is no topical or transdermal commercial product of tramadol HCl available in the market.Therefore,a comparative anti-inflammatory effect was investigated with commercial tramadol HCl tablet,F4,placebo proniosomes and negative control.Fig.5 compares the percentage of edema after treatment in various groups.It is apparent from Fig.5 that the data of group II and group IV are very distinct as compared to negative control (group I) and placebo treated (group III).Among the drug administered groups,certainly transdermal therapy of F4 showed prominent inhibition of induced inflammation or edema (P < 0.05)when compared to oral therapy of marketed formulation(group II).Indeed,the percentage of edema (0.67%-7.52%)was low and consistent throughout the study period (8 h),implies the transdermal application of F4 improved the clinical efficacy of tramadol.The improvement in clinical efficacy of transdermal therapy of tramadol can be correlated with the pharmacokinetic data observed with transdermal administration of F4 (group IV) and oral therapy (group II)(Table 4).It is apparent from the Table 4 that theAUC0-8 hvalues observed in the transdermal therapy of F4 was significantly (P <0.005) higher than the oral administration.In addition,the greaterCmax(56.02 ± 6.05 μg/ml) and delayedTmax(4 h) in the transdermal therapy of F4 may likely toincrease the clinical efficacy and extend the therapeutic effect as compared to its oral counterpart.Followed by transdermal therapy,the positive control (marketed tablet administered by oral route) showed certain inhibition of inflammation.The percentage of edema by the oral therapy of tramadol with similar dose was 6.78%-11.18% (until 8 h),signifies its moderate efficiency and also correlate with theAUC0-8 hvalues observed in the Table 4.In contrast,both negative control and placebo group’s shown higher edema in rats and were similar and not statistically significant.The percentage of edema observed in control and placebo group’s varied from 16.38% to 33.12% during the study period (8 h).Overall,the data observed here indicate the prospective of F4 to rapidly transport niosome into and through the skin and deliver tramadol for systemic effect.

Table 3-Stability data of proniosome gel(F4)at different conditions.
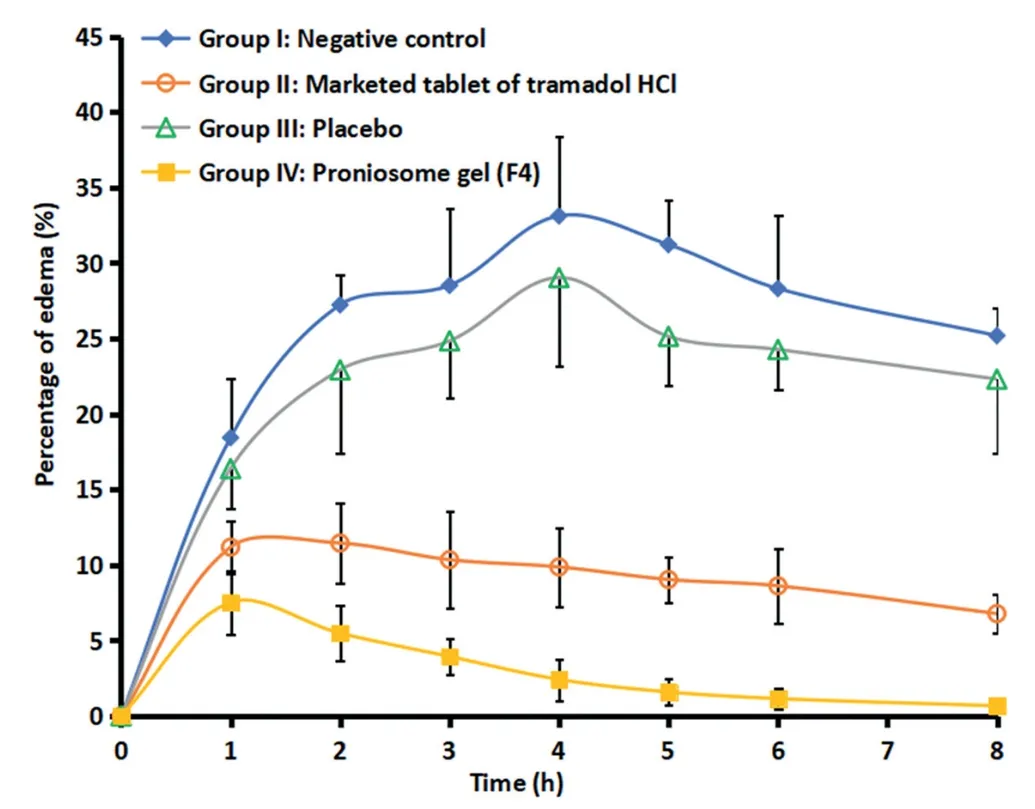
Fig.5-Comparison of mean inhibition of edema in hind paw of rats in various treatments.The profiles between negative control and placebo resemble each other and are statistically insignificant.On the other hand,the edema profile of treated group(proniosome gel,F4)is statistically different as compared to other treatments(negative control,placebo and positive control groups)at P <0.005.The data represents average±SD(n=6).

Table 4- Mean pharmacokinetic parameters of tramadol in plasma after transdermal delivery of selected proniosome gel (F4) (group IV) and oral administration(group II)in rats(n=6).
3.10.Antinociceptive activity
Antinociceptive studies with various treatments signify that the number of writhes observed in mice decreases as;negative control (64.78 ± 3.02)>marketed tablet (25.45 ± 2.01)>F4(20.67 ± 1.78).In addition,the number of writhes observed with F4 was significantly different (P <0.005) as compared to oral marketed tablets.Moreover,F4 significantly decreased the number of twitches,by 68.09%,in comparison to the oral marketed tablet(P <0.05),which also reduced the chemically induced writhes over 60.71%.Plasma drug concentration in mice was measured at 30,45,60,and 75 min after dose administration in group II (oral marketed tablet) and group III (F4) in order to assess the possible correlation between tramadol plasma level and antinociceptive effect.Indeed,theAUC0-1.25 hvalue of F4(20.29±4.12 μg·h/ml)and oral marketed tablet (13.26 ± 3.50 μg·h/ml) were significantly different(P <0.01),and correlated with the observed antinociceptive effect.Therefore,it was established that the transdermal therapy of tramadol using F4 has more efficacy than its oral counterpart.
3.11.Skin irritation
Skin irritancy study was performed to determine the probable localized skin sensitivity of F4 and control because skin safety is an important concern for transdermal products.The F4 showed a Draize dermal scoring grade value of “0’’,thus confirming to be as non-sensitive to human skin [29].No noticeable erythema,edema or inflammation was visible on rabbits’skin after seven days of topical use of F4.
4.Conclusion
It is evident from present investigation that the F4 contributes the highest EE%.Furthermore,F4 provides greater transdermal flux with~5 folds enhancement in comparison to control.A significant improvement of anti-inflammatory(P <0.05)and antinociceptive(P <0.005)effects was witnessed from F4 in comparison to marketed oral tramadol tablets and is correlated to pharmacokinetic data.Typically,proniosome vesicular systems produce depot in the deeper skin layers and continuously release the drug over time,which is advantageous as it reduces the application frequency and provides sustain therapy in the management of chronic pain.In general,these findings conclude that proniosome gel formulation can be successfully used to improve the clinical efficacy of tramadol.Furthermore,the transdermal therapy of tramadol could be more advantageous as it overcomes various issues associated with oral administration including opioid-like effects and first pass metabolism.Incorporation of chemical skin permeation agents may further improve the bioavailability of tramadol from this novel formulation,F4,via transdermal route.
Conflicts of interest
The authors report no conflicts of interest.
Acknowledgment
The authors are highly thankful to Arihant School of Pharmacy &BRI,Gandhinagar and Institute of Pharmacy,Nirma University,Ahmedabad for providing laboratory facilities.
REFERENCES
[1]Skinner DJ,Epstein J,Pappagallo M.Tramadol.In:Smith HS,editor.Current therapy in pain.Philadelphia:Elsevier;2009.p.508-12.
[2]Mattia C,Coluzzi F.Tramadol:a wonder drug for the treatment of chronic pain?Int J Clin Rheumatol 2010;5(1):1-4.
[3]Vazzana M,Andreani T,Fangueiro J,Faggio C,Silva C,Santini A,et al.Tramadol hydrochloride:pharmacokinetics,pharmacodynamics,adverse side effects,co-administration of drugs and new drug delivery systems.Biomed Pharmacother 2015;70(C):234-8.
[4]Epstein DH,Preston KL,Jasinski DR.Abuse liability,behavioral pharmacology,and physical-dependence potential of opioids in humans and laboratory animals:lessons from tramadol.Biol Psychol 2006;73(1):90-9.
[5]WHO,Tramadol pre-review report.https://www.who.int/medicines/access/controlled-substances/PreReview_Tramadol.pdf [Accessed on 12 September 2018].
[6]Grond S,Radbruch L,Lehmann KA.Clinical pharmacokinetics of transdermal opioids:focus on transdermal fentanyl.Clin Pharmacokinet 2000;38:59-89.
[7]Nair AB,Jacob S,Al-Dhubiab BE,Alhumam RN.Influence of skin permeation enhancers on the transdermal delivery of palonosetron:an in vitroevaluation.J Appl Biomed 2018;16(3):192-7.
[8]Marwah H,Garg T,Goyal AK,Rath G.Permeation enhancer strategies in transdermal drug delivery.Drug Deliv 2016;23(2):564-78.
[9]Raffa RB.Basic pharmacology relevant to drug abuse assessment:tramadol as example.J Clin Pharm Ther 2008;33:101-8.
[10]Ammar HO,Ghorab M,El-Nahhas SA,Kamel R.Polymeric matrix system for prolonged delivery of tramadol hydrochloride,part I:physicochemical evaluation.AAPS PharmSciTech 2009;10:7-20.
[11]Anroop B,Ghosh B,Parcha V,Khanam J.Transdermal delivery of atenolol:effect of prodrugs and iontophoresis.Curr Drug Deliv 2009;6(3):280-90.
[12]Takasuga S,Yamamoto R,Mafune S,Sutoh C,Kominami K,Yoshida Y,et al.In vitroandin vivotransdermal iontophoretic delivery of tramadol,a centrally acting analgesic.J Pharm Pharmacol 2011;63(11):1437-45.
[13]Kamboj S,Bala S,Nair AB.Solid lipid nanoparticles:an effective lipid based technology for poorly water soluble drugs.Intl J Pharm Sci Rev Res 2010;5(2):78-90.
[14]Neubert RHH.Potentials of new nanocarriers for dermal and transdermal drug delivery.Eur J Pharm Biopharm 2011;77(1):1-2.
[15]Khatoon M,Shah KU,Din FU,Shah SU,Rehman AU,Dilawar N,et al.Proniosomes derived niosomes:recent advancements in drug delivery and targeting.Drug Deliv 2017;24(2):56-69.
[16]Ahmad MZ,Mohammed AA,Mokhtar Ibrahim M.Technology overview and drug delivery application of proniosome.Pharm Dev Technol 2017;22(3):302-11.
[17]Jacob S,Nair AB,Al-Dhubiab BE.Preparation and evaluation of niosome gel containing acyclovir for enhanced dermal deposition.J Liposome Res 2017;27(4):283-92.
[18]Nobilis M,Kopecký J,Kvĕtina J,Chladek J,Svoboda Z,Voříšek V,et al.High-performance liquid chromatographic determination of tramadol and its O-desmethylated metabolite in blood plasma:application to a bioequivalence study in humans.J Chromatogr A 2002;949(1-2):11-22.
[19]Al-Dhubiab BE,Nair AB,Kumria R,Attimarad M,Harsha S.Formulation and evaluation of nano based drug delivery system for the buccal delivery of acyclovir.Colloids Surf B Biointerfaces 2015;136:878-84.
[20]Nair AB,Sammeta SM,Vaka SRK,Murthy SN.A study on the effect of inorganic salts in transungual drug delivery of terbinafine.J Pharm Pharmacol 2009;61(4):431-7.
[21]Nair A,Gupta R,Vasanti S.In vitrocontrolled release of alfuzosin hydrochloride using HPMC-based matrix tablets and its comparison with marketed product.Pharm Dev Technol 2007;12(6):621-5.
[22]Nair A,Reddy C,Jacob S.Delivery of a classical antihypertensive agent through the skin by chemical enhancers and iontophoresis.Skin Res Technol 2009;15(2):187-94.
[23]Anroop B,Ghosh B,Parcha V,Kumar A,Khanam J.Synthesis and comparative skin permeability of atenolol and propranolol esters.J Drug Del Sci Tech 2005;15(2):187-90.
[24]Thakur R,Anwer MK,Shams MS,Ali A,Khar RK,Shakeel F,et al.Proniosomal transdermal therapeutic system of losartan potassium:development and pharmacokinetic evaluation.J Drug Target 2009;17(6):442-9.
[25]Jacob S,Shirwaikar A,Nair A.Preparation and evaluation of fast-disintegrating effervescent tablets of glibenclamide.Drug Dev Ind Pharm 2009;35(3):321-8.
[26]Nair A,Morsy MA,Jacob S.Dose translation between laboratory animals and human in preclinical and clinical phases of drug development.Drug Dev Res 2018;79(8):373-82.
[27]Adzu B,Amos S,Wambebe C,Gamaniel K.Antinociceptive activity of Zizyphus spina-christi root bark extract.Fitoterapia 2001;72(4):344-50.
[28]Young HY,Luo YL,Cheng HY,Hsieh WC,Liao JC,Peng WH.Analgesic and anti-inflammatory activities of[6]-gingerol.J Ethnopharmacol 2005;96(1-2):207-10.
[29]Wilhelmus KR.The Draize eye test.Surv Ophthalmol 2001;45(6):493-515.
[30]Shah H,Nair AB,Shah J,Bharadia P,Al-Dhubiab BE.Proniosomal gel for transdermal delivery of lornoxicam:optimization using factorial design andin vivoevaluation in rats.DARU J Pharm Sci 2019.doi:10.1007/s40199-019-00242-x.
[31]Kumar GP,Rajeshwarrao P.Nonionic surfactant vesicular systems for effective drug delivery-an overview.Acta Pharm Sin B 2011;1(4):208-19.
[32]El-Laithy HM,Shoukry O,Mahran LG.Novel sugar esters proniosomes for transdermal delivery of vinpocetine:preclinical and clinical studies.Eur J Pharm Biopharm 2011;77(1):43-55.
[33]Yoshioka T,Sternberg B,Florence AT.Preparation and properties of vesicles(niosomes)of sorbitan monoesters(Span20,40,60 and 80)and a sorbitan triester(Span85).Int J Pharm 1994;105(1):1-6.
[34]Essa E.Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes.Asian J Pharm 2010;4(4):227-33.
[35]Bayindir ZS,Yuksel N.Characterization of niosomes prepared with various nonionic surfactants for paclitaxel oral delivery.J Pharm Sci 2010;99(4):2049-60.
[36]Yusuf M,Sharma V,Pathak K.Nanovesicles for transdermal delivery of felodipine:development,characterization,and pharmacokinetics.Int J Pharma Investig 2014;4(3):119-30.
[37]Bansal S,Aggarwal G,Chandel P,Harikumar SL.Design and development of cefdinir niosomes for oral delivery.J Pharm Bioallied Sci 2013;5(4):318-25.
[38]Aboelwafa AA,Doaa AE,Aliaa NE.Comparative study on the effects of some polyoxyethylene alkyl ether and sorbitan fatty acid ester surfactants on the performance of transdermal carvedilol proniosomal gel using experimental design.AAPS PharmSciTech 2010;11:1591-602.
[39]Fang JY,Yu SY,Wu PC,Huang YB,Tsai YH.In vitroskin permeation of estradiol from various proniosomes formulation.Int J Pharm 2001;215:91-9.
[40]Lamana SMS,Napimoga MH,Nascimento APC,Freitas FF,de Araujo DR,Quinteiro MS,et al.The anti-inflammatory effect of tramadol in the temporomandibular joint of rats.Eur J Pharmacol 2017;807:82-90.
 Asian Journal of Pharmacentical Sciences2020年6期
Asian Journal of Pharmacentical Sciences2020年6期
- Asian Journal of Pharmacentical Sciences的其它文章
- Recent trends on wound management:New therapeutic choices based on polymeric carriers
- Evolution from small molecule to nano-drug delivery systems:An emerging approach for cancer therapy of ursolic acid
- BioPerine Encapsulated Nanoformulation for Overcoming Drug-Resistant Breast Cancers
- NIR-triggered thermo-responsive biodegradable hydrogel with combination of photothermal and thermodynamic therapy for hypoxic tumor
- Dynamic micelles with detachable PEGylation at tumoral extracellular pH for enhanced chemotherapy
- Regorafenib-loaded poly(lactide-co-glycolide)microspheres designed to improve transarterial chemoembolization therapy for hepatocellular carcinoma