
泛基因组研究在遗传多样性和功能基因组学中的应用
2021-01-12 01:00向坤莉贺文闯邹益彭丹张晓妮廖雪竹王杰杨健康武志强
广西植物 2021年10期
关键词:遗传多样性
向坤莉 贺文闯 邹益 彭丹 张晓妮 廖雪竹 王杰 杨健康 武志强

摘 要:相对于单个参考基因组仅聚焦于个体遗传信息的挖掘,泛基因组研究则能够反映整个物种或类群全部的遗传信息。随着基因组测序和分析技术的不断发展,泛基因组学逐渐成为新的研究热点,并已在植物、动物和微生物多个物种中获得了广泛应用,为全面解析物种或类群水平的遗传变异和多样性、功能基因组和系统进化重建等研究提供了强有力的工具,取得了很多显著的研究成果。尽管如此,由于泛基因组学研究尚处于发展阶段,测序费用和分析成本仍然较高,难以广泛普及;且存在分析标准不一、数据挖掘不够全面深入、理论难以应用于生产实际等尚待解决的问题,仍有较大的发展空间。该文系统总结了泛基因组在生物遗传多样性挖掘和功能基因组学中的研究进展,主要包括其在泛基因组图谱的构建、基因组变异和有利基因的发掘、功能基因的多态性、群体遗传多样性和系统进化等多个领域中的应用和研究,探讨了其在不同领域的应用潜力。同时,讨论了目前泛基因组研究中存在的局限性和可能的解决方法,并对其将来的发展前景进行了展望。
关键词:泛基因组,结构变异,功能基因,遗传多样性,系统进化
中图分类号:Q943.2
文献标识码:A
文章编号:1000-3142(2021)10-1674-09
Abstract:The pan-genome can represent all of the genetic diversities in a species or population,which is a limitation for obtaining only one single reference genome. The pan-genomics is becoming a new hot research area and being widely applicated in researches of many species in plants,animals and microorganisms,as the development of the whole genome sequencing and analysis technology. It provides powerful tools for resolving the genetic variation and polymorphism at levels of species or taxa,researches of functional genomics and reconstruction of phylogenetics,obtaining abundant of significant research achievements. However,present researches on pan-genomics still need to improve due to several problems,e.g.,extensive cost of whole genome sequencing and data analysis,inconsistent analysis standards,lack of deeper and comprehensive explanation of the obtained data,and difficulty of application of the research achievements. We summarized the research progresses of pan-genomes on exploitation of genetic diversity and functional genomics,including construction of a pan-genome map,identification of genome variations and favorable genes,polymorphism of functional genes,population genetic diversity and systematic evolution,and discussed its potential in application of different research fields. Furthermore,we discussed the limitations existed in the present studies and possible solutions,and presented the prospect in the future on pan-genomics.
Key words:pan-genome,structural variants,functional gene,genetic diversity,systematic evolution
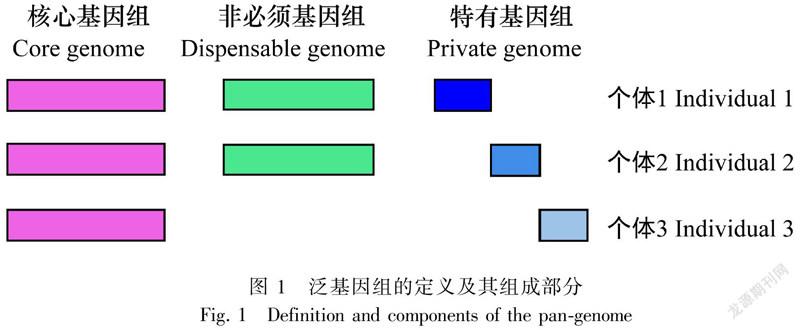
遺传变异是生物进化的内在源泉,因而,研究遗传多样性及其演化规律是生物遗传学及进化生物学研究中的核心问题之一。而泛基因组研究则是近年来随着测序成本的急剧降低和分析技术的快速发展而全面反映物种遗传变异的一种新兴工具。泛基因组研究能够从物种或类群水平广泛发掘和利用遗传变异多样性,是现代医学、生物学、农学中的一个前沿领域。其中,泛基因组(pan-genome)指一个物种或者类群的全部基因组信息的集合,包括核心基因组(core genome)和非必须基因组(dispensable genome)两部分。核心基因指在所有个体中都存在的基因/组分集合;而非必须基因组是指在部分个体或单个个体中存在的基因/组分集合,有时也称为可变基因组(variable genome) (图1; Tettelin et al.,2005; Medini et al.,2005)。核心基因组由所有样本中都存在的序列组成,往往与重要的生物学功能和表型特征相关,多数是一些管家基因(house-keeping genes),反映了物种的稳定性;可变基因组由仅在部分样本中存在的序列组成,一般与物种对特定环境的适应性或特有的生物学特征相关,反映了物种的多样性和特异性(Montenegro et al.,2017; Gordon et al.,2017; Wang et al.,2018; Zhao et al.,2018; Liu et al.,2020)。
当前,泛基因组研究已经广泛应用于多个植物、动物和微生物物种中,为全面解析物种或类群水平的遗传变异、功能基因研究和系统进化重建等研究提供了强有力的工具,取得了很多显著的研究成果(付静和秦启伟,2012; 王娅丽等,2019; Tian et al.,2019; Chen et al.,2020; Domínguez et al.,2020; Weissensteiner et al.,2020; Liu et al.,2020)。然而,现有的泛基因组学研究主要聚焦于不同个体基因组序列和基因结构的变异(Montenegro et al.,2017; Zhao et al.,2018; Gao et al.,2019; Liu et al.,2020),但对这些变异如何介导基因结构和功能的改变,最终影响生物表型,以及这种遗传差异如何与环境因子互作,都未能进行深入探讨。本文综述了泛基因组学在不同物种中的研究进展,对其在群体基因组变异、功能基因的鉴定和发掘、群体遗传多样性和系统进化等多个领域中的应用与研究进行了系统性总结,并对其应用前景和局限性进行了探讨。
1 泛基因组图谱的构建
最早在2005年,Tettelin et al.(2005)在对几种链球菌属细菌(GBS,group B Streptococcus)的遗传多样性研究中提出微生物的泛基因组概念,指出核心基因组是在所有菌株中都存在的基因;非必须基因组(可变基因组)是仅在部分菌株中存在的基因。其中GBS菌共有的核心基因组占80%,剩余20%的基因组信息为非必须基因组。随后,2010年Li et al. (2010)通过对多个人类个体基因组进行组装和比较基因组学分析,提出了“人类泛基因组”的概念,也就是人类群体基因组信息的总和,并从中鉴定获得新发现的序列达到19~40 Mb。而随着千人基因组计划的提出和实施,泛基因组在人类疾病方面的研究取得了许多重大突破,为精准医疗计划提供了可能(1 000 Genomes Project Consortium,2012)。
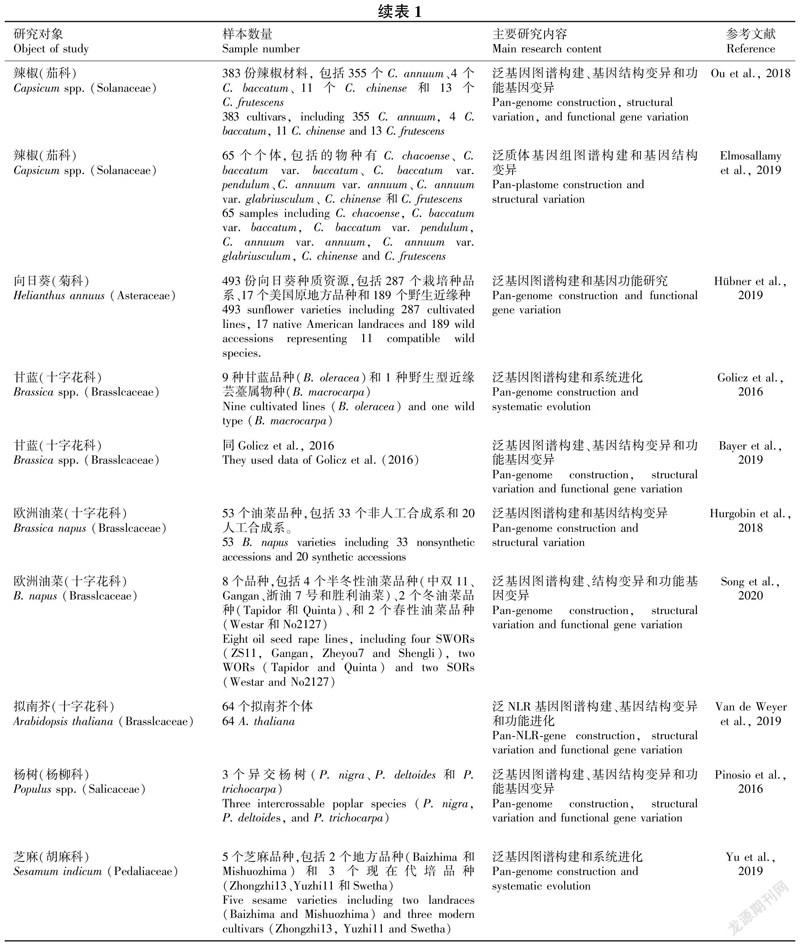
之后,随着越来越多的物种完成了高质量基因组参考序列的组装,多个动植物物种中相继报道了泛基因组图谱的构建相关研究工作。例如,通过对全球12个种猪品种的基因组进行高质量组装,构建了猪的泛基因组图谱,发现中国的猪品种有大约9 Mb的泛序列(pan-sequences)与欧洲的猪品种存在差异,其中包括脂肪细胞脂解的必要调节因子TIG3 (Tazarotene-induced gene 3)(Tian et al.,2019);對19个小麦品种的泛基因组分析发现,平均每个样本中含有128 656个基因,核心基因有89 795个(Montenegro et al.,2017);利用725个番茄品种的基因组信息构建的番茄泛基因组图谱中,整个番茄泛基因组共包含40 396个基因,其中74.2%是核心基因(Gao et al.,2019)。此外,泛基因组在水稻(Schatz et al.,2014;Yao et al.,2015; Sun et al.,2017; Wang et al.,2018; Zhao et al.,2018; Zhou et al.,2020)、大豆(Li et al.,2014; Liu et al.,2020; 祝光涛和黄三文,2020)、玉米(Hufford et al.,2012; Hirsch et al.,2014; 简银巧等,2017)等重要的植物物种均获得了广泛应用(表1)。因此,构建整个物种的泛基因组图谱已成为广泛应用的基因组学方法,不仅能够发现全面的遗传信息,而且能为从物种和群体水平进行功能基因组学、系统进化和其他生物学研究提供更强有力的工具。
2 泛基因组学研究中序列结构变
异与功能基因发掘
同一物种内一个或几个参考基因组能够反映的遗传变异是非常有限的,而泛基因组研究能够覆盖物种或类群中的所有变异,为研究整个物种或类群水平上的基因组序列和结构变异提供了可能。现代生物基因库中的遗传变异通常包括单核苷酸多态性(SNPs,single-nucleotide polymorphisms)、插入缺失(Indels,insertions and deletions)和大的结构变异(SVs,large structural variants)。其中SVs主要包括拷贝数变异(CNVs,copy number variants)、存在/缺失变异(PAVs,presence/absence variants)、移位(translocation events)和倒置(inversion events)等,而这些变异往往和一些关键的农艺性状相关(Springer et al.,2009; Hirsch et al.,2014; Li et al.,2014; Lu et al.,2015; Zhao et al.,2018)。
通过泛基因组分析全面发掘群体基因组中的序列和结构变异,能够鉴定其中与有利表型相关的变异位点,为发掘和研究新的功能基因提供了重要依据。例如,利用 66个水稻高质量基因组构建了水稻的泛基因组,从中共鉴定到16 563 789 个SNPs、5 549 290个Indels和933 489个SVs,分析了其中与开花时间相关的基因Hd3a (Heading date 3a)、抗寒性基因COLD1 (Chilling tolerance divergence 1)、谷物重量基因GW6a (Grain weight 6a)、分蘖角度基因TAC1 (Tiller Angle Control 1)、植株高度基因Sd1 (Semi dwarf 1)在不同材料间的遗传变异,表明SNPs变异是导致这些基因变异的基础(Zhao et al.,2018)。而利用29个高质量基因组构建的大豆泛基因组图谱,共鉴定获得14 604 953个SNPs、12 716 823个Indels和776 399个SV(包含723 862个PAVs、27 531个CNVs、21 886个移位和3 120个倒置),发现有些结构变异在重要农艺性状调控中发挥重要作用,如PAV、基因融合和Indels分别对种皮亮度、种皮颜色的驯化、缺铁失绿等性状具有重要影响(Liu et al.,2020)。
同时,在不同层次上发现的多个序列和结构变异,不仅提供了更加丰富的变异信息,也为研究基因功能变异提供了更多素材。例如,通过六倍体普通小麦物种基因组间和亚基因组间的共線性分析,提出其“4A-5A-7B染色体重排”是两次染色体易位事件的结果,并明确了重排的基因组区间的精细边界;并且在微观尺度上探讨了小麦春化基因Vrn2 (Vernalization2)的复杂进化历史,发现Vrn2同源基因在普通小麦基因组中的复杂分布是包含串联重复、多倍化、染色体易位和基因丢失在内的一系列事件叠加的结果(Chen et al.,2020)。另有研究利用100个番茄基因组捕获到238 490个SVs,构建得到泛结构变异(panSV)图谱,研究表明SVs是许多转座子的基础,而且SVs集中区域的基因渐渗现象严重,且群体中90%的SVs变异可在泛基因组图谱中获得验证(Alonge et al.,2020)。
3 泛基因组学研究中功能基因的
变异与多态性
遗传结构变异通常会导致基因功能的改变,泛基因组研究能够通过全面整合相关基因的遗传信息,揭示基因重组、融合等事件导致基因功能的获得、丢失,以及发掘新基因。例如,大豆缺铁萎黄病有关的候选基因被定位于14号染色体上,通过泛基因组研究发现该候选基因有两种单倍型:品种“中黄13”所属的单倍型主要分布在低纬度地区;品种“威廉82”所属的单倍型主要分布在高纬度地区,能够在高pH值、铁为不易吸收的难溶氧化物等环境中生存,这种单倍型启动子区有1.4 kb 的Indel和外显子区有5个变异位点(Liu et al.,2020)。在油菜中通过全PAV-GWAS (genome wide association study)分析发现3个开花抑制因子BnaA10.FLC、BnaA02.FLC和BnaC02.FLC的PAVs与油菜的开花时间和生态型分化密切相关,其中:冬油菜品种的BnaA10.FLC启动子区都含有MITE (miniature inverted repeat transposable element)插入;85%春油菜品种的BnaA10.FLC第一个外显子中含有LINE (long interspersed nuclear elements)插入;81%半冬性油菜品种的BnaA10.FLC启动子区含有hAT插入。表明BnaA10.FLC决定了油菜生态类型,是控制油菜开花的关键基因(Song et al.,2020)。
生物的表型往往是来自多个基因网络调控的结果,其中很多基因可能又同时对多个不同的表型性状具有影响,因此对某个表型的有利基因亦有可能对另一个表型具有不利影响。例如,现代番茄中的产量相关性状调控机制复杂,对100个番茄基因组的泛结构变异(pan-SV)的研究发现,由四个结构变异导致形成了三个MADS-Box基因,共同影响番茄的经济性状。其中j2TE基因型具有便于收获的无关节花梗表型,而ej2w基因型具有防止撞伤的大花萼表型,但两个基因型同时存在(j2TE ej2w)则会出现花序分枝过多而导致低育性的现象;sb1(suppressor of branching 1)基因型能有效克服双隐性基因型的负面作用,实现增产;另外,sb1基因型的表达可能受1号染色体上STM3基因的串联重复序列影响,且串联重复的拷贝数具有剂量效应 (Alonge et al.,2020)。因此,通过在更广泛的群体中研究基因功能变异对表型的影响,将有助于更加准确地对功能基因-表型的关联做出全面详细地评估,从而更好地指导分子育种工作来培育出抗病性更强、产量更高、保质期更长、风味更好的作物品种,同时又不牺牲其他所期望的表型性状。作物泛基因组学研究已经发现了大量农艺表型与特定基因的存在、缺失和变异之间的多样化的相关性(Tao et al.,2019),通过在泛基因组完整遗传图谱的基础上进行研究,将有利于彻底澄清其内在关联和相应的机理。
4 泛基因组学研究在种群遗传多样性和系统进化研究中的应用
对泛基因组学的研究,不仅可以全面地从基因组水平分析物种内遗传多样性,探究个体间的系统发生关系和表型差异的遗传基础,而且可以从物种、亚种水平分析基因组的序列变异和系统进化特征,为研究物种的起源及演化等重要生物学问题提供依据。例如,通过水稻泛基因组对6个水稻群体中与驯化有关的7个基因位点开展进化分析,发现Aus群体(Indica的一个亚类群)并未全部聚在栽培稻进化分支上,从而提出Aus水稻群体处于不完全驯化选择状态(Zhao et al.,2018)。利用小麦泛基因组对19个小麦个体基因的PAVs进行了发掘并构建了系统进化树,发现小麦品种‘中国春’位于进化树的基部,为小麦不同类型种质的系统进化关系和研究利用提供了理论依据(Montenegro et al.,2017)。对32只乌鸦群体的泛基因组研究,将鸦属(Corvus)分为Jackdaw和Crow两大支系,并在此基础上探讨了不同进化分支上乌鸦的基因组结构变异和功能性状,尤其是发现乌鸦羽毛图案差异大,但遗传差异不大,主要受NDP 基因上游20 kb处一个大小为2.25 kb的LTR (long terminal repeats)逆转座子插入调控 (Weissensteiner et al.,2020)。
泛基因组研究还可运用于对不同生态地理类型中差异较大的种质资源进行基因组测序,挖掘物种中新的基因,为候选基因的补充、物种多样性及适应性进化、起源经历和外来物种入侵性等问题的研究提供重要信息。例如,大豆群体的生物地理分析发现现代栽培大豆起源于中国的华北地区(Liu et al.,2020),而水稻群体的相关研究发现现代栽培稻起源地应该包括中国华南地区(Huang et al.,2012)。此外,由于一些作物的基因库中包括多个物种,特别是具有不同遗传结构的野生近缘物种,需要构建含该作物所有品种及其近缘种的遗传图谱以进行更广泛的研究,因此也有学者提出了超-泛基因组(super-pan-genome)的概念,以探讨更大范围种质群体的遗传基础及其多样性(Khan et al.,2020)。
5 泛基因组学研究的发展前景
真核生物的全部基因组信息包括核基因组、线粒体基因组和质体基因组。目前的泛基因组学研究大多关注的是核基因组,而线粒體和质体这两种细胞器的泛基因组研究也逐渐开始被重视。例如,研究者利用PCAWG (The Pan-Cancer Analysis of Whole Genomes) 数据库中2 658个癌症样本及其匹配的正常组织样本的全基因组数据构建了人类线粒体基因组最全面的突变蓝图,并确定了多个高度突变类型,其中截断突变(truncated mutations)在肾脏癌症、结直肠癌和甲状腺癌中明显富集,提示了激活特殊的信号通路或会带来致癌影响(Yuan et al.,2020)。此外,有研究者利用321个辣椒的叶绿体基因组,构建了辣椒5个栽培种及2个变种的叶绿体泛基因组,其不但用系统发育信号分析揭示了辣椒属不同种间亲缘关系的远近,也对7个叶绿体泛基因组的CDS (coding sequence)、内含子和基因间隔区的遗传多样性进行了详尽分析,确定了rpl23和trnI的基因间隔区包含44 bp串联重复以及其他插入缺失和单核苷酸等丰富的变异(Elmosallamy et al.,2019)。
在某些物种中,由于其基因组较大和可移动元件的比例较高等原因,使得泛基因组研究难以有效开展,因此,关注全部RNA信息的泛转录组(pan-transcriptome)研究开始逐渐兴起。许多重要作物,如玉米(Hansey et al.,2012; Hirsch et al.,2014; 简银巧等,2017)和大麦(Ma et al.,2019),以及模式生物拟南芥(Gan et al.,2011)等的泛转录组研究均已有报道。
随着多种测序技术的结合和分析策略的发展,泛基因组学相关研究呈现爆发式增长,但是大多数研究的深入程度不一,许多数据结果仍有进一步深入挖掘的空间。尤其是构建完整的基因图谱后,很多研究止步于对某几个基因的结构变异进行鉴定,未进一步开展系统的功能研究,更不用说应用于生产实践。此外,随着大量生物信息学数据的积累,单个团队面对浩大的数据库也只能选择部分数据结果进行深入研究,难以充分利用现有的数据。例如,人类基因组计划从开始启动到现在已经过去30年,仍需大量的人力投入和研究分析去解决更多的问题。因而,完善的数据共享机制和良好平台是泛基因组学研究良性发展和应用的一个重要条件。目前,我国已建立了国家基因组科学数据中心(NGDC,National Genomics Data Center),某些重要农作物或农业动物物种的泛基因组数据也建立了数据分享平台,如猪的泛基因组数据库PIGPAN (http://animal.nwsuaf.edu.cn/code/index.php/pan-Pig)、大白菜基因组数据库BRAD (the Brassica database,http://brassicadb.cn)和油菜泛基因组资源数据库(http://cbi.hzau.edu.cn/bnapus/)等。
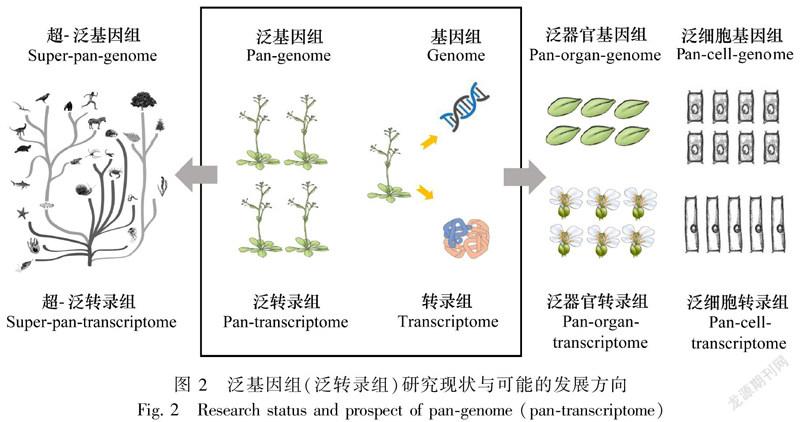
一方面,进一步整合更广泛的多层次群体基因组数据,如不同世代之间的泛基因组研究、整合多个物种的超-泛基因组研究等,可能是值得进一步探索的新方向(图2)。另一方面,随着测序技术的不断发展,尤其是单细胞测序技术的发展和测序成本的进一步降低,单细胞分辨率的转录组图谱已经逐步开始在水稻和玉米的根发育研究中获得应用(Satterlee et al.,2020; Liu et al.,2021)。因此,同一个体不同组织器官的泛基因组或泛转录基因组研究,乃至不同细胞之间的泛基因组或泛转录基因组研究也可能成为新的发展方向(图2)。
参考文献:
1000 GENOMES PROJECT CONSORTIUM,2012. An integratedmap of genetic variation from 1092 human genomes [J]. Nature,491(7422):56-65.
ALONGE M,WANG X,BENOIT M,et al.,2020. Major impacts of widespread structural variation on gene expression and crop improvement in tomato [J]. Cell,182(1):145-1161.
BAYER PE,GOLICZ AA,TIRNAZ S,et al.,2019. Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome [J]. Plant Biotechnol J,17(4):789-800.
CHEN YM,SONG WJ,XIE XM,et al.,2020. A collinearity-incorporating homology inference strategy for connecting emerging assemblies in Triticeae tribe as a pilot practice in the plant pangenomic era [J]. Mol Plant,13(12):1694-1708.
DOMNGUEZ M,DUGAS E,BENCHOUAIA M,et al.,2020. The impact of transposable elements on tomato diversity [J]. Nat Commun,11(1):4058.
ELMOSALLAMY MM,OU LJ,YU HY,et al.,2019. Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species [J]. Hort Res,6(1):108.
FU J,QIN QW,2012. Pan-genomics analysis of 30 Escherichia coli genomes [J]. Hereditas,34(6):765-772. [付静,秦启伟,2012.30株大肠杆菌的泛基因组学特征分析[J]. 遗传,34(6):765-772.]
GAN X,STEGLE O,BEHR J,et al.,2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana [J]. Nature,477:419-423.
GAO L,GONDA I,SUN H,et al.,2019. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor [J]. Nat Genet,51(Suppl.):1044-1051.
GOLICZ AA,BAYER PE,BARKER GC,et al.,2016. The pangenome of an agronomically important crop plant Brassica oleracea [J]. Nat Commun,7(1):13390.
GORDON SP,CONTRERAS-MOREIRA B,WOODS DP,et al.,2017. Extensive gene content variation in the Brachypodium distachyon pangenome correlates with population structure [J]. Nat Commun,8(1):2184.
HANSEY CN,VAILLANCOURT B,SEKHON RS,et al.,2012. Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing [J]. PLoS ONE,7:e33071.
HIRSCH CN,FOERSTER JM,JOHNSON JM,et al.,2014. Insights into the maize pan-genome and pan-transcriptome [J]. Plant Cell,26(1):121-135.
HUANG XH,KURATA N,WEI XH,et al.,2012. A map of rice genome variation reveals the origin of cultivated rice [J]. Nature,490(7421):497-501.
HBNER S,BERCOVICH N,TODESCO M,et al.,2019. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance [J]. Nat Plants,5(1):54-62.
HUFFORD MB,XU X,VAN HEERWAARDEN J,et al.,2012. Comparative population genomics of maize domestication and improvement [J]. Nat Genet,44:808-811.
HURGOBIN B,GOLICZ AA,BAYER PE,et al.,2018.Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus [J]. Plant Biotechnol J,16(7):1265-1274.
JAYAKODI M,PADMARASU S,HABERER G,et al.,2020. The barley pan-genome reveals the hidden legacy of mutation breeding [J]. Nature,588(7837):284-289.
JIAN YQ,2017. Variations in pan-transcriptome and genome size in tropocal Maize (Zea mays L.) and their applications [D]. Beijing:Chinese Academy of Agricultural Sciences. [簡银巧,2017. 热带玉米全长泛转录组和基因组大小变异及应用[D]. 北京:中国农业科学院. ]
KHAN AW,GARG V,ROORKIWAL M,et al.,2020. Super-pangenome by integrating the wild side of a species for accelerated crop improvement [J]. Trends Plant Sci,25(2):148-158.
LI RQ,LI YR,ZHENG HC,et al.,2010. Building the sequence map of the human pan-genome [J]. Nat Biotechnol,28:57-63.
LI YH,ZHOU GY,MA JX,et al.,2014. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits [J]. Nat Biotechnol,32(10):1045-1052.
LIU Q,LIANG Z,FENG D,et al.,2021. Transcriptional landscape of rice roots at the single cellrsolution [J]. Mol Plant,14(3):384-394.
LIU YC,DU HL,LI PC,et al.,2020. Pan-genome of wild and cultivated soybeans [J]. Cell,182(1):162-176.
LU F,ROMAY MC,GLAUBITZ JC,et al.,2015. High-resolution genetic mapping of maize pan-genome sequence anchors [J]. Nat Commun,6:6914.
MA YL,LIU M,STILLER J,et al.,2019. A pan-transcriptome analysis shows that disease resistance genes have undergone more selection pressure during barley domestication [J]. BMC Genomics,20:12.
MABIRE C,DUARTE J,DARRACQ A,et al.,2019. High throughput genotyping of structural variations in a complex plant genome using an original Affymetrix Axiom array Supplementary figures and tables [J]. BMC Genomics,20:848.
MEDINI D,DONATI C,TETTELIN H,et al.,2005. The microbial pan-genome [J]. Curr Opin Genet Dev,15(6):589-594.
MONTENEGRO JD,GOLICZ A,BAYER PE,et al.,2017. The pangenome of hexaploid bread wheat [J]. Plant J,90(5):1007-1013.
OU LJ,LI D,LV JH,et al.,2018. Pan-genome of cultivated pepper (Capsicum) and its use in gene presence-absence variation analyses [J]. New Phytol,220(2):360-363.
PINOSIO S,GIACOMELLO S,FAIVRE-RAMPANT P,et al.,2016. Characterization of the poplar pan-genome by genome-wide identification of structural variation [J]. Mol Biol Evol,33(10):2706-2719.
SATTERLEE JW,STRABLE J,SCANLON MJ,2020. Plant stem cell organization and differentiation at single-cell resolution [J]. Proc Natl Acad Sci USA,117:33689-33699.
SCHATZ MC,MARON LG,STEIN JC,et al.,2014. Whole genome de novo assemblies of three divergent strains of rice,Oryza sativa,document novel gene space of aus and indica [J]. Genome Biol,15:506.
SONG JM,GUAN ZL,HU JL,et al.,2020. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus [J]. Nat Plants,6(1):34-45.
SPRINGER NM,YING K,FU Y,et al.,2009. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content [J]. PLoS Genet,5(11):e1000734.
SUN C,HU ZQ,ZHENG TQ,et al.,2017. RPAN:rice pan-genome browser for approximately 3000 rice genomes [J]. Nucl Acids Res,45(2):597-605.
TAO YF,ZHAO XR,MACE E,et al.,2019. Exploring and exploiting pan-genomics for crop improvement [J]. Mol Plant,12(2):156-169.
TETTELIN H,MASIGNANI V,CIESLEWICZ MJ,et al.,2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae:Implications for the microbial “pan-genome” [J]. Proc Natl Acad Sci USA,102(39):13950-13955.
TIAN XM,LI R,FU WW,et al.,2020. Building a sequence map of the pig pan-genome from multiple de novo assemblies and Hi-C data [J]. Sci Chin Life Sci,63(5):750-763.
VAN DE WEYER AL,MONTEIRO F,et al.,2019. A species-wide inventory of NLR genes and alleles in Arabidopsis thaliana [J]. Cell,178(5):1260-1272.
WALKOWIAK S,GAO L,MONAT C,et al.,2020. Multiple wheat genomes reveal global variation in modern breeding [J]. Nature,588(7837):277-283.
WANG WS,MAULEON R,HU ZQ,et al.,2018. Genomic variation in 3,010 diverse accessions of Asian cultivated rice [J]. Nature,557(7703):43-49.
WANG YL,ZHU SS,YANG FS,et al.,2019. Pan-genome sequencing and comparative genomic analysis of atrazine-degrading bacteria [J]. Biotechnol Bull,35(7):90-99. [王娅丽,朱姗姗,杨峰山,等,2019. 莠去津降解菌泛基因组测序及比较基因组分析 [J]. 生物技术通报,35(7):90-99. ]
WEISSENSTEINER MH,BUNIKIS I,CATALN A,et al.,2020. Discovery and population genomics of structural variation ina songbird genus [J]. Nat Commun,11(1):3403.
YAO W,LI GW,ZHAO H,et al.,2015. Exploring the rice dispensable genome using a metagenome-like assembly strategy [J]. Genom Biol,16(1):187.
YU JY,GOLICZ AA,LU K,et al.,2019. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars [J]. Plant Biotechnol J,17:881-892.
YUAN Y,JU YS,KIM Y,et al.,2020. Comprehensive molecular characterization of mitochondrial genomes in human cancers [J]. Nat Genet,52:342-352.
ZHAO Q,FENG Q,LU HY,et al.,2018. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice [J]. Nat Genet,50:278.
ZHOU Y,CHEBOTAROV D,KUDRNA D,et al.,2020. A platinum standard pan-genome resource that represents the population structure of Asian rice [J]. Sci Data,7:113.
ZHU GT,HUANG SW,2020. A 360-degree scanning of population genetic variations—A pan-genome study of soybean [J]. Chin Bull Bot,55(41):56-65. [祝光濤,黄三文,2020. 360度群体遗传变异扫描——大豆泛基因组研究[J]. 植物学报,55(41):403-406. ]
(责任编辑 周翠鸣)
猜你喜欢
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
长江蔬菜·学术版(2016年12期)2017-01-12
河北渔业(2016年9期)2016-11-05
天津农业科学(2015年5期)2015-05-30
天津农业科学(2015年5期)2015-05-30
江苏农业科学(2015年1期)2015-04-17
