
天然胶体的水环境行为*
2021-01-12 11:04:24王沛芳包天力
湖泊科学 2021年1期
王沛芳 ,包天力,胡 斌,钱 进
(1:河海大学浅水湖泊综合治理与资源开发教育部重点实验室,南京210098) (2:河海大学环境学院,南京210098)
胶体是一种形态分散的混合物,广泛地存在于河湖、地下水、海洋等自然水体之中[1-3]. 胶体具有丰富的官能团,对有机类以及疏水性污染物有着良好的吸附能力,胶体表面的电荷则可以通过静电产生吸附效应. 被胶体结合后,污染物在胶体的表面可以发生多种物理化学以及生物过程,例如溶解沉淀、氧化还原、离子交换、光化学降解和生物降解等等[1,4-6]. 因此,胶体的固-液界面被认为影响着水环境中大量活性物质的浓度与循环过程. 此外,作为一种纳米级颗粒,胶体在水环境中难以直接沉降,其絮凝沉降的过程以及对水动力条件的响应与一般的悬浮颗粒物相差巨大,从而对污染物的迁移转化过程有着更加复杂的影响[3, 7-8]. 近年来,关于胶体对污染物的吸附、输移作用备受关注,然而在天然水体中,较低的浓度使得胶体难以提取和准确检测,因此在过去的研究中,胶体被笼统地划入溶解态中,导致胶体的环境作用未能得到充分的认识[3];但随着超滤技术、场流分离技术等分离技术与检测技术的发展,对天然胶体进行深入研究的条件已经成熟. 本文基于近年来的研究成果,总结了水环境中胶体概念的引入与发展,讨论比较了多种胶体的提取、表征手段,归纳总结了胶体对污染物的环境作用,进而指出当前研究中,胶体组分内有机质的作用尚未得到足够的重视,胶体态污染物的毒理特性与胶体形态、组成的动态变化对胶体性质、环境行为影响的研究较为不足.
1 水环境研究中胶体概念的引入与发展
物质在水环境中的赋存形态,影响着其反应活性与环境行为,区分物质在水环境中的形态,特别是区分物质的溶解态与固态是水环境研究的基础之一[6]. 1952年,Goldberg使用0.5 μm孔径的醋酸纤维滤膜对海水中的颗粒态物质进行了分离,并逐渐形成了如今被广泛应用的以0.45 μm滤膜分离溶解-颗粒态物质的习惯[9-12]. 然而,基于溶解-颗粒二相体系的吸附动力学推论与实际观测结果间却存在较大的分歧,例如,吸附实验中,重金属从溶解相吸附到固体颗粒中的速度被发现低于理论值[13];吸附过程中存在 “颗粒浓度效应(particle concentration effect)”,即在吸附实验中,随着颗粒物浓度的上升,吸附物在溶解态、颗粒态之间的分配系数Kd出现明显下降的现象[14-16];以及地下水研究中,钚等放射性元素的实际迁移距离被发现远远大于基于溶解-颗粒二相模型的计算值[17-19].
为了准确地描述物质在水体中复杂的分布与迁移行为,胶体的概念被引入水环境研究中. 1989年Honeyman和Santschi提出了“胶体泵”理论,强调重金属等污染物能够先被胶体快速吸附,再随胶体逐渐团聚形成大型颗粒,解决了重金属实际吸附速度低于理论值的矛盾[13]. 另一方面, 胶体概念的引入也为“颗粒浓度效应”提供了一种解释:在较高的颗粒物浓度下,胶体对溶质的吸附量上升,增强了传统溶解态的吸附能力,从而导致了吸附物分配系数下降[13,15,20]. 1999年,Benoit和Rozan发现,如果将胶体从传统的溶解态物质中分离,则分配系数将不随颗粒浓度生变化[21]. 这些发现表明,在研究水体中的污染物,尤其是那些受到固-液表面性质影响的物质时,将胶体从传统溶解态物质中独立出来是极为必要的.
胶体概念的引入为研究水环境中物质的输移转化提供了新的视角,并取得了一系列研究成果. 例如,海洋学研究发现“海洋雪”中的有机物大部分是胶体形态,并认为胶体态有机质在溶解性有机质(DOM)与颗粒态有机质(POM)的转化中起到中介作用[3];Wu等2001年在Science上发表文章,认为海洋中的“溶解”铁很大一部分以胶体的形式存在[10],胶体的聚集与沉降对海洋的物质循环具有重要意义. 在地下水研究方向,许多研究证实了多孔介质中胶体能够促进重金属的迁移[20,22-23],并总结了pH值、胶体浓度、离子强度等因素的具体影响[24-26].
近年来,胶体的研究得到了进一步发展,河湖水体中胶体的作用得到进一步重视,胶体对污染物分布与迁移转化的影响从传统的重金属与氮、磷逐渐扩展到新型有机污染物、微生物等领域,胶体的研究对象也由实验室内人工制备的单一成分胶体转向环境中组成复杂的天然胶体,多个研究已经开始探索环境中普遍的氧化还原、光化学反应中胶体的作用. 当前,环境中的微型颗粒物被广泛关注,研究水环境中的胶体将有利于进一步完善水环境中物质循环体系,控制各类污染物的环境危害.
2 胶体的定义、分类与来源
2.1 胶体的定义
胶体的粒径通常被定义为1 nm~1 μm[6]. 然而,由于水环境研究中通常以0.45 μm滤膜分离水体中的颗粒物,因此许多研究将胶体定义为粒径为1 nm~0.45 μm的非溶解物质[14,27-30]. 目前,各类研究普遍采用超滤技术来排除1 nm以下的溶解态物质,因超滤膜、超滤装置的孔径通常以分子量为单位,所以通常以1 kDa代替1 nm作为胶体尺寸的下界(值得注意的是,物质的分子量为1 kDa接近但不完全等同于物质的粒径为1 nm). 目前各类研究对胶体粒径的定义并不统一,其中部分研究采用了1 nm~1 μm的经典胶体定义[23,31-33],也有研究根据自身实验器材限制以及实验目的差异,选择3 kDa、10 kDa等较小的分子量作为胶体体积的下界.
单纯以粒径判别胶体缺乏理论上的根据,也使得采用不同粒径范围的研究之间难以相互对照. 因此,不少研究者试图从化学性质与环境意义的角度对胶体进行认识与定义. 这些研究集中强调了胶体必须具有固体类型的表面性质和特殊的水动力特性,认为与溶解态相比,胶体在体积上应大于溶解态物质,其内部结构及其表面性质应具有固体特性或异于水溶液,从而有利于从溶液中吸附溶质;与颗粒态相比,胶体受重力沉降、孔隙阻拦的作用微弱,必须通过聚集形成大颗粒后才能够沉降. 在这种定义中,胶体与溶解态、颗粒态物质的粒径范围将存在重叠部分,而纤维素等有机大分子尽管拥有符合胶体定义的分子量,但因为缺乏吸附疏水物质的能力,也将不属于胶体物质的范畴[3].
从表面性质与水动力特性的角度对胶体物质进行认知有助于研究者理解胶体在环境中的反应与作用,但尚不能就胶体的获取、鉴别提出便捷可行的操作方案,因此,以粒径定义胶体依然是水环境中胶体研究的主流方法. 同时,天然水体中的胶体往往与溶解态、颗粒态物质共同存在,其浓度也低于实验室人工制备的各种胶体溶液,因此,目前尚无手段能够全面地直接检测、表征水环境中的胶体,必须通过各种以粒径为指标的分离技术对胶体进行分离、提取[11].
2.2 胶体的分类与来源
在使用粒径作为胶体的判定标准时,水环境中胶体的组分极为复杂,可能包括微生物、生物碎片、有机质、各种矿物黏土颗粒、金属的氧化水解产物,甚至可能包括微塑料以及人工纳米材料的环境次级产物[8,34-36]. 根据组成与来源,胶体可分为包含纳米材料、微塑料、生物炭、石墨烯等因人类活动而进入环境的人工胶体,以及水环境自身变化产生的天然胶体. 为了避免学科间的重复研究,不少学者对胶体的组成进行了适当简化,着重强调在水环境中最为普遍的那一类胶体,即由Fe、Mn、Al等金属的氧化物、水化物或矿物黏土形成的无机亚微米颗粒以及胶体态有机质共同组成的分散物质[34-37].
此外,例如病毒、细菌等微生物在环境中也拥有符合胶体定义的粒径,因此在部分研究中也被称为“生物胶体”. 与传统的非生物胶体相比,生物胶体不存在溶解与颗粒形态,且具有生命体特有的繁殖、代谢、对外界刺激产生反应等生物特性,因此其环境作用迥异于传统胶体. 由于微生物在孔隙介质中的迁移规律与胶体具有相似之处,如具有团聚-分散特性,受到静电作用影响等,因此,在环境研究中,“生物胶体”的概念目前主要集中在关注微生物迁移行为的相关研究中.
天然水体中的胶体来自于胶体的组成成分在环境中的赋存以及特定条件下的“胶体化”. 除了微塑料、纳米材料等因人类活动而进入水环境的特殊胶体以外,胶体的无机来源主要为各类土壤、沉积物以及悬浮颗粒物,有机来源主要为生物活动产生并在环境中积存的有机质.
胶体在水体中拥有较强的迁移能力,从其他水体中输入是水环境中胶体的重要来源之一,而胶体在环境中的原位产生机制主要有以下几类:
1)水动力条件变化导致的胶体生成. 通过水流变化,水体能够在土壤、沉积物等固体介质表面形成切向力场,当水动力作用超过颗粒物间的黏合力时,将导致固定在固体介质上的胶体重新释放到水体中,同时也导致一部分土壤、沉积物破碎解离,形成新的胶体颗粒. 水动力促进胶体生成的现象广泛地发生在土壤、沉积物中,在降水下渗、再悬浮等水力条件显著变化的环境过程中尤其显著. 例如,Zhang 等在长江上游地区实地研究发现,强降雨期间地下径流中的胶体质量浓度峰值可达到188 mg/L[38];Xu在浅水湖泊再悬浮现象的研究中发现,当风速由2 m/s上升到8 m/s时,太湖中的胶体浓度由20.4 mg/L上升到58.4 mg/L,沉积物再悬浮过程对太湖水体中胶体的贡献率高达55%[39].
2)水化学环境变化导致的胶体生成. 包括溶液的离子强度、pH值、氧化还原程度以及有机质在内的多种水化学要素均能影响胶体粒子的相互作用力,影响胶体的生成与去除. 通常,离子强度下降能够通过增加胶体表面的电势来增强胶体间的斥力,从而有利于形成胶体. 因此,部分研究提出了胶体间电势能垒为零时的临界盐浓度(critical salt concentration ,CSC)以及临界离子强度(critical total ionic strength,CTIS)来描述胶体的产生过程,当溶液的盐浓度、离子强度低于临界浓度时,水环境中的胶体将出现显著的动员现象[40]. 例如,在地下水研究中,当电导率低于50 μS/cm的水下渗到电导率为150 μS/cm的含水层时,胶体在地下水中出现了明显的动员现象,达到了30 mg/L的浓度[20]. 除了重新活化已经固定的胶体颗粒,pH值的上升以及氧化作用都能使水体中的Fe、Mn、Al等离子形成其氧化或氢氧化物,并逐渐团聚形成胶体,有文献指出,在富含腐殖酸的环境下,被氧化的Fe(Ⅱ)离子能够与腐殖酸共同形成主要粒径小于200 nm的复合胶体[41].
3)生物活动产生的胶体. 在天然水体中,微生物、动植物的生命代谢持续消耗、生产着各类有机质,而陆地有机质也能够通过降雨径流、渗流、淹没等方式输送到水环境中. 这些有机质中一部分直接以胶体的形式存在于水环境之中,如藻类产生的胞外聚合物(EPS),而溶解态或颗粒态的有机质也能够通过聚集和溶解的方式转变为胶体态. 此外,部分研究也将环境中的各类微生物称为“生物胶体”.
3 天然水体中胶体的分离与表征
3.1 胶体的分离技术
目前,以粒径定义胶体依然是水环境中天然胶体研究的主流方法,而胶体的分离、提取,也主要基于各种以粒径为指标的分离技术,其中较为常用的有超滤技术(UF)、流场分离技术(FFF)、尺寸排除色谱法(SEC)以及基于胶体沉降特性的沉降法(表1).
3.1.1 超滤技术 超滤技术是当前运用最为广泛的胶体分离手段. 超滤能够以回流液的形式回收体积大于滤膜孔径的物质,适用于提取胶体等拥有最小粒径限制的物质. 超滤技术已经较为成熟,具有原理简单、体积小、易于操作等诸多优点,并发展出切向超滤、离心超滤等多个类型.
超滤技术的主要缺点在于难以将物质以粒径进行细致的划分,而不同粒径的胶体在物质构成与性质上存在差异,根据粒径对胶体进行进一步的细分是极有必要的[42-44]. 此外,有研究指出超滤膜对物质的截留能力与物质粒径的大小呈曲线关系,超出其标定孔径的物质仍有一定概率穿过滤膜[45]. 超滤较低的粒径控制能力限制了其在粒径精确度要求较高的研究中的应用. 其次,超滤对胶体的细分程度受限于超滤膜的数量,在需要分离出多个粒径范围的胶体时,必须使用多种孔径的超滤膜联立成多级过滤装置,在增加系统的复杂程度与操作成本的同时也降低了超滤的准确性.
基于质量守恒的回收率是评估超滤效果的重要指标,能否保证过滤后回收率保持在较高水平是衡量超滤技术应用是否成功的关键. 回收率越高,则超滤的效果越好,相关实验数据的精度也越高. 除了回收率以外,胶体的粒径变化也是超滤过程中值得注意的事项. 在超滤过程中经过加压、混合、过膜后,胶体的粒径会产生一定的变化,并根据超滤技术的具体类型而存在差异. 在垂直超滤、切向超滤以及离心超滤3种方式中,垂直超滤被观测到能够导致胶体团聚,增大胶体的平均尺寸,而切向、离心两种方式的影响则较为微弱[46]. 因此,在应用超滤技术时,应审慎选择超滤膜的材质、孔径、超滤的具体技术形式并控制超滤过程中的时长、压力、流速等因素,以减小超滤过程对胶体物质的干扰.
3.1.2 场流分离技术 场流分离是一种常用于生物、纳米材料等领域的分离技术. 场流分离技术的原理是将含有待提取物质的液体通过一道细长的分离通道,由于分离通道具有极高的宽高比,因此通道内液体的流动接近二维层流,在这种情况下,接近通道壁的流速几乎为零,而通道中部的流速最快. 通道内垂直施加的力场使得样品朝向底部积累,由于底部浓度的提高而产生反向的扩散力,驱使分析物返回到通道中央. 当扩散力与垂直力场作用平衡时,分析物将在垂直方位上静止平衡,从而处于不同的流速之中以达到分离物质的目的[47-48]. 其中典型的场包括横向流场、温度场、电势场、磁场等等,因此场流分离技术能够根据分析物在尺寸、热扩散、电荷、磁化率等性质上的差异进行物质分离.
在胶体以及纳米材料研究中,应用最为广泛的为流场流分离装置(FlFFF),该装置通过垂直的流场提供作用力,在处理尺寸1 μm以内的分析物时,可以将胶体按照尺寸大小进行连续分离,通过与ICP-MS、荧光分光光度计、紫外分光光度计等检测设备联用,能够完整地分析胶体在整个粒径尺度上的性质差异. 同时,场流分离技术对胶体形态影响较小,在流场条件一致的情况下,对胶体的分离效果不受器材差异的影响,避免了各实验之间因超滤膜材质型号不同带来的差异. 由于分离效果好,实验精度高,场流分离装置在胶体研究领域有着广阔的应用前景[42,49-50].
场流分离技术的缺点是仪器价格昂贵且处理量少,样品在分离过程中必须与载体溶液混合,只适用于制备少量样品进行相关指标的检测,难以获取大批量的胶体.
3.1.3 尺寸排阻色谱 尺寸排阻色谱通过色谱技术以分离不同分子量的物质[42,51-52]. 在尺寸排阻色谱中,低分子量物质能获得更多的内部孔隙体积,而大分子物质则被排除在孔隙之外. 因此,大分子量物质会首先排出,然后是分子量较小的部分. 尺寸排阻色谱分离胶体物质时的干扰由胶体组分与色谱柱的填料间的相互作用产生. 在洗脱过程中,胶体物质可能与色谱图填充材料间产生斥力,导致部分分子以比实际分子量更高的表面分子质量排出,也有可能被电荷吸引,从而被吸附在色谱柱填料内.
3.1.4 沉降法 沉降法根据胶体的沉降速度区分胶体. 悬浮颗粒在静止液体中的沉降速度取决于它们各自的大小、形状和密度. 因此,根据斯托克斯公式,可计算具有一定粒径的胶体在静水中的沉降速度,结合溶液的静止时间,可大致得出该粒径胶体的沉降深度,而此深度以上的溶液中则只包含小于该粒径的颗粒[39]. 此外,针对胶体沉降速度较慢的特点,部分研究则利用离心机产生的离心力加速胶体粒子的沉降,通过控制转速与时间,能够促使特定粒径以上的胶体颗粒加速沉降,从而达到分离胶体的目的. 然而,现实中的颗粒往往具有非均质性,其沉降速度不一,导致沉降法的精度较差;其次,胶体的沉降速度较慢,耗时较长,不利于快速展开实验.

表1 胶体分离技术以及特点对比
3.2 胶体的表征技术
胶体是一类混合物,胶体复杂的组分决定了其表征手段一方面要体现胶体的含量、结构、粒径、总体电势等胶体的普遍特性,另一方面要能针对胶体中某一类组分的特殊性质进行表征,因此,胶体的表征往往要结合多种技术手段.
就如何表征胶体这一混合物总量的问题,有学者提出了一系列公式,这些公式根据胶体中有机碳的含量以及典型的矿物元素的浓度(如Al、Fe等),乘以相应的经验参数折算成胶体的总量[53]. 显然,特定的公式仅适用于组成较为固定的特定胶体,并不具有普适性. 此外,也有研究将胶体浓缩液冷冻干燥后的干重作为胶体总量的表征,将进一步去除有机物后的干重作为无机胶体总量的表征,将二者的质量差用以表征有机胶体总量[39]. 胶体的结构、粒径通常使用电镜以及其他光学技术进行表征,其中,电镜图像是微观物质研究中最为直观有效的手段,扫描电子显微镜(SEM)、透射电子显微镜(TEM)等获得的胶体图像能够清晰地表现出无机胶体以及一部分有机胶体的形态特征,可用于尺寸、形状和结构的分析;静态光散射(SLS)与动态光散射(DLS)技术利用溶液中悬浮颗粒散射形成的光强与角度的函数关系,能够获得胶体粒度大小与形状的数据,而采用激光衍射技术(LD)的粒度分析仪通常用于纳米材料表征,也能够表征胶体的粒径以及数量. 此外,胶体的总体电势一般通过测量胶体的zeta电位进行表征,可通过各类zeta电位仪实现.
在通用型的表征手段以外,胶体研究往往需要针对胶体某单独组分的性质进行表征,依据胶体组分的不同,这类表征技术大体可分为有机和无机两类.
水环境中的有机胶体通常由天然有机质(NOM)组成. 其中,溶解性有机质(DOM)被定义为粒径<0.45 μm的有机质组分,而有色可溶解性有机质(CDOM)则定义为其中的显色部分,CDOM能强烈吸收紫外线等光线,可以通过其光学特性来评估DOM的相对水平[54-56]. 由于有着与胶体相近的粒径范围,用于溶解性有机质性质研究的技术手段都可以运用于有机胶体的表征.

通过这些技术手段,当前研究已经对有机胶体的含量与性质进行了分析. 表2列举了一些代表性河湖水体中胶体有机碳含量在总溶解态中的比重,表明了在总量上,胶体有机质在溶解性有机质中占有较为重要地位[64-65].

表2 部分水环境中胶体有机碳在总溶解态有机碳中的比例
有机胶体的含量及性质也随着分子量发生变化. 流场流分离技术的结果表明,绝大部分的溶解性有机质分布在100 kDa以内的胶体与真溶解态中[70-71]. 有机质在胶体内的集中使得有机胶体具有突出的化学性质,包括紫外-可见光吸收度、荧光强度在内的多个光学指标,均在1~10 nm/kDa的胶体物质内出现显著的峰值,表明小型有机胶体具有更强的芳香性,可能是DOM中主要的活性物质[72]. 不同类型荧光有机质的分布也被发现与分子量存在密切的关联,Cuss 和 Gueguen发现类蛋白荧光组分分布在<1 kDa的真溶解态中,微生物类腐殖质和陆生类腐殖质主要集中在2 kDa左右[73]. Romera-Castillo则通过尺寸排阻色谱得到了相似的结果:类腐殖质组分主要富集在中等分子量范围内 (5 kDa左右),而蛋白质类组分的分布变化较大,分别分布在分子量最高和最低的组分中[52,74].
胶体的无机部分主要由各类黏土颗粒、金属氧化物组成,其粒径大小、形态结构可以通过胶体通用的表征手段进行分析. 无机胶体中的元素组成可以在无机胶体进行酸化后,通过电感耦合等离子体原子发射光谱(ICP-AES)或质谱(ICP-MS)直接检测胶体中相关元素的浓度;此外,例如能量色散谱仪(EDS)、X射线衍射光谱(XRD)则可以进一步检测胶体颗粒的元素构成与晶体结构,为无机胶体颗粒的研究提供更多的信息[40,75-76]. 此外,BET法也可用于测定无机胶体的比表面积以及孔容,并进一步表征胶体颗粒的吸附性能.
相对于有机胶体,无机胶体在性质上较为稳定. 基于多种检测手段,许多研究成功实现了对无机胶体的表征. Wang利用电镜、能谱技术在对地下水胶体粒子进行了研究,观测到无机胶体的平均直径和高度分别为322 nm ± 90 nm和10.4 nm ± 2.1 nm,其矿物学组成主要为钠长石、正长石、斜绿泥石、纤铁矿、白云母、方解石和石英[76]. Xu 也通过类似方法研究了太湖中无机胶体的组成,发现无机胶体中的主要元素为Si、Fe、Al、K和Mg,其矿物组成为石英和蒙脱石,主要粒径分布为40~200 nm[32].
4 胶体与水环境中的污染物
4.1 胶体中污染物的含量与分布
胶体对营养盐、重金属、抗生素、激素等新型有机污染物均有着较强的结合能力,是它们在水体中的重要存在形态. 胶体中污染物的含量与分布,胶体与污染物的结合能力以及机理,是这一方向的重点研究内容.
4.1.1 胶体中的氮、磷 胶体是水体中营养盐的重要载体. 有研究在苏格兰北部5条河流中发现,胶体态总氮平均占溶解态的25%,其中有机氮占溶解态的比例为58%,无机氮占溶解态的比例仅为7%. 由于无机氮的主要成分以离子形式存在,故胶体氮中的无机氮含量极少,主要组成为有机氮. 在土壤中,胶体态氮的比例被发现与总氮含量呈负相关,在低氮含量土壤中,0.45 μm以内超过75%的氮为胶体态,而在高氮含量土壤中,胶体态的比重下降为42%,表明胶体态可能是环境中氮的优先结合形态[77]. 对于磷,研究发现胶体中总磷平均占溶解态的比重为42%,其中钼酸盐活性磷占比为34%,有机磷占比为59%[14]. 此外,Van Moorleghem等在比利时河流中发现胶体态磷占据了溶解态磷含量的52%[78],Gottselig等通过对欧洲范围内96个河流断面的胶体进行分析,发现胶体磷的平均比例为50%,波动范围为36%~70%[79],王芳等在太湖、千岛湖等5类典型水体中发现胶体态可酶解磷占总磷的比例为16.6%~55.6%[80].
除了有机结合态的磷以外,胶体中无机结合态的磷被认为是水体中磷的重要组成部分[81]. 胶体对磷的吸附作用主要来自其中的铁基胶体. 水体中铁的水解产物通常对磷有着很强的吸附作用,而胶体铁是水体中铁(氢)氧化物的重要组成部分. 胶体对磷的吸附与磷铁之间的摩尔比(P∶Fe)存在密切的关联,在低磷铁比时,磷主要以胶体态存在;随着P∶Fe值上升,胶体对磷的吸附出现饱和,磷的主要赋存形式转变为真溶解态,并导致胶体磷所占比例下降. Stijn在比利时的5条河流里检测了胶体中铁与磷的含量,发现P∶Fe值为0.02~1.5时,磷主要以铁胶体结合态存在,在P∶Fe值更高的情况下以真溶解态存在. 在解吸实验中,饱和吸附产生的胶体磷绝大部分能够7天内从胶体中释放,而低磷浓度下胶体与磷的结合体则非常稳定,表明了不同磷浓度下,胶体对磷的结合机理以及形成的结合物存在着较大差异[82-83]. 有研究认为,除去表面吸附外,铁胶体还可能通过与磷形成羟基磷酸铁的方式增强与磷的结合[84]. 胶体的结合不但改变了磷在水体中的分布,也影响了磷的稳定性以及生物可利用性,天然的含磷胶体中的磷不能直接被生物利用,减少了由游离态磷带来的富营养化风险[85-86].
4.1.2 胶体中的新型有机污染物 胶体被认为是抗生素等新型有机污染物(EOCs)的重要载体,许多研究已经从胶体内检测到了较高浓度的抗生素、激素等有机污染物. Cheng等使用超滤技术从白洋淀湖水中研究了抗生素在胶体中的分布,发现溶解性抗生素平均浓度为13.65~320.44 ng/L,其中胶体态约占4.7%~49.8%,表明胶体是水生环境中重要的抗生素载体[87];晏彩霞对长江口如抗生素、环境内分泌干扰物等42种EOCs的研究也发现胶体具有较高的EOCs浓度,对EOCs总浓度的贡献率可达42.3%. 通过检测真溶解态、胶体态、颗粒态中的EOCs浓度,EOCs在胶体中的分配系数比颗粒态高出1~2个数量级,表明了胶体对EOCs强烈的吸附作用[88]. 目前,胶体对EOCs吸附的机制尚处于初步阶段,不少研究已经发现胶体态EOCs浓度与胶体碳之间的正相关关系[89]. 场流分离的结果显示,1~5 kDa的胶体(即有机胶体的主要分布区间)对EOCs的吸附能力最强,随着粒径的增加,胶体中EOCs浓度不断下降,可以认为胶体中的有机组分是吸附EOCs的主要成分. 胶体对EOCs的吸附也受到环境因素的影响,比如盐度、溶液离子强度的升高能显著降低胶体对EOCs的吸附[90].
4.1.3 胶体中的重金属 重金属是胶体研究中最受关注的一类污染物,胶体具有的带电性、比表面积巨大等特性被认为能够有效地结合水体中的重金属. 1996年,Wen等使用超滤装置从海水中提取出胶体并检测了其中重金属的含量,发现溶解态中65%~85%的Cu、35%~40%的Ni以及30~95%的Pb以胶体的形式存在[11]. 其后,Wen等在加尔维斯顿湾三一河的实验数据则显示,溶解态中约45%±9%的Cd、55%±4%的Cu、19%±6%的Co、36%±6%的Ni、64%±9%的Pb、91%±5%的Zn以及79%±11%的Fe为胶体态[91]. 也有研究在10 kDa~0.2 μm的粒径区间内检测到海水中胶体态Al、Ag和Fe在总溶解态中占比将近84%,胶体态Cu和Mn的比例则为16%~20%,而Zn和Ni的比重为2%~3%[92]. 在河湖水体中,张战平等以1 kDa~1 μm为胶体的粒径范围,研究了太湖典型湖区胶体态痕量金属含量及其季节变化,总结出太湖春季水体中胶体态Cu、Zn、Pb、Fe、Mn和Cd平均占据各自总溶解态的22.3%、26.3%、20.7%、58.8%、40.4%和59.2%[93-95]. 黄文丹对长江口水体中胶体重金属的研究数据显示,胶体态Fe、Cu、Mn、Ni、Zn和As等种重金属占总溶解态的比例超过50%[68]. 重金属在胶体中的含量表明,水环境中的胶体在重金属的环境行为中具有重要的作用.
不同胶体组分对重金属的结合之间存在显著的差异. 通过相关性分析,部分重金属在胶体中的含量与胶体有机碳的浓度相关,从而推测这部分重金属在胶体中可能以金属-有机质络合物的形式存在,而Fe和Mn(以及部分研究中的Al、Pb)等重金属含量则与胶体有机碳浓度的相关性较弱. 利用场流分离技术,Stolpe等在密西西比河以及墨西哥湾的水体中发现大部分重金属元素结合在0.5~40 nm的胶体中,其中例如Cu、Cr、As等重金属主要结合在3~8 nm的有机胶体中,而5~40 nm区间内的无机胶体内主要含有Mn、Zn和Pb等重金属[50]. 黄文丹通过多级超滤发现长江口水体中,超过65%的胶体态Fe、Mn分布在大分子量胶体(>500 kDa)中,而超过50%的胶体态Pb和As分布在小分子量胶体中(<500 kDa)[68]. 根据重金属在不同分子量胶体内的含量,可将胶体与重金属的结合大致归结为3个类型,分别为:结合在有机胶体区间的重金属(如Cu和Cr)、结合在无机胶体区间的重金属(如Fe和Mn)以及部分混合类的重金属(如Zn 和Hg).
4.2 胶体对污染物的吸附机制
胶体对水体中各种微量污染物如重金属等有着强烈的吸附能力,其最主要的吸附能力被认为来源于胶体的带电性[96]. 由于胶体粒子表面的电离以及胶体对溶液中部分离子存在选择性吸附,大多数胶体都携带着一定数量的电荷,例如胶体表面官能团(例如羟基)的酸碱反应以及表面离子的吸附/解吸作用均能产生电荷. 以金属氧化物为代表的胶体颗粒为例,其表面与水溶液接触时会水解形成羟基,并随着pH值变化产生正电荷或是负电荷[6].
除了通过静电吸引来吸附与胶体表面电荷相反的带电离子外,胶体还可以通过其他方式吸附各类污染物,例如范德华力、氢键作用、配体交换或是螯合作用形成化学键进行吸附. 目前,这类发生在界面上的吸附行为通常被称为表面络合反应,并在1970年由Schindler和Stumm提出了表面络合模型(SCM)用于描述[97]. 表面络合模型认为固体颗粒的表面存在着大量的官能团,可以通过形成配位键与水体中的溶质发生络合反应,进而将溶质吸附到固体的表面. 其中,羟基被认为是胶体颗粒表面最重要的官能团之一,通过在表面结合配位水,胶体无机颗粒中的金属氧化物和氢氧化物能够通过水解反应形成拥有大量羟基的表面.
以羟基与金属离子结合为例,在反应平衡受溶液pH值的影响下,有:
(1)
(2)
[ST]=[SOH]+[SOM]
(3)
代入后有:
(4)
以吸附量ΓM代替[SOM]后,可化为Langmuir吸附方程式的形式:
(5)
目前,胶体吸附的研究主要应用以Langmuir和Freundlich为代表的吸附等温线模型,主要关注的污染物也集中在重金属一类. 相关研究发现,高岭土胶体对Pb、Cu等重金属的吸附,氧化铁胶体对天然有机质的吸附,以及湖底沉积物制备的胶体溶液对Pb的吸附都可用Langmuir等温线模拟[22,28,32]. Painter以钚为示踪剂,用蒙脱石、赤铁矿、二氧化硅和蒙皂石配置了无机胶体溶液,吸附实验的结果表明无机胶体的吸附行为基本符合二级动力学模型[98]. 对于有机胶体,则主要通过荧光光谱的淬灭现象以及Ryan-Weber模型进行表征. Ryan-Weber模型建立在有机质官能团与金属离子一比一形成配合物的假设上,并构建了荧光强度与有机质官能团间的关系. 具体应用上,如Wu分析了海水中不同分子量有机质与Cu(Ⅱ)的结合能力,发现胶体态的结合系数较高,与重金属结合的能力较强[10]. 大部分有机胶体与金属离子的淬灭实验中,胶体态有机质的结合能力强于传统的溶解态,部分强于“真溶解态”的有机质[99-101].
对于其他物质,Yoon等研究了磷与氧化铁胶体溶液的吸附行为,发现氧化铁对磷酸盐的吸附在酸性条件下保持稳定,在碱性条件下大幅下降,吸附行为为自发吸热过程,符合伪二级动力学模型[102];Lu等通过吸附试验发现胶体在Fe、Mn氧化物表面的吸附也符合二级动力学模型,通过Langmuir方程估算出在pH为5时,每摩尔Fe对磷的吸附率为0.223 mol,并进一步将吸附归因于静电作用和通过形成单配位和双配位配合物取代氧化物的表面羟基[103]. 此外,也有研究发现,胶体对雌激素的吸附符合Langmuir模型,推测胶体对雌激素的吸附为单层吸附,成分不同的胶体对雌酮和雌二醇的最大吸附量分别能够达到90.09~3333和116.28~2000 mg/kg[104].
这些成果表明,对于单一组分的胶体,经验吸附模型能够较好地揭示其吸附规律,但在组分混合的情况下,规律将变得复杂. 胶体有机质已经被发现能显著加强Al2O3等纳米颗粒以及矿物胶体对重金属离子的吸附[105]. 有机质能够与无机胶体、重金属离子通过静电作用、配体交换、氢键或是疏水作用等关系发生复杂的结合[106]. 如图1所示,有机质本身足以与无机胶体以复杂的方式形成表面吸附. 胶体的有机组分与无机颗粒形成复合体后,将改变胶体的结构、比表面积、表面电荷和表面官能团等性质,进而影响胶体的吸附能力;根据复合体的形态,胶体的无机、有机组分间既有可能形成点位掩蔽,减少污染物质的吸附,也有可能形成新的吸附点位,增强胶体的吸附能力. 此外,有机质在矿物胶体表面的吸附还可以增强抑或抑制矿物的溶解,取决于具体有机质与矿物的吸附机理以及溶液成分[107],并能够促使金属-矿物配体在饱和溶解度条件下,由颗粒态转变为胶体[108]. 对于非金属类污染物,有机质则有可能与同样带有负电的污染物竞争无机胶体上的结合点位,如土壤有机质被发现与磷酸盐在针铁矿、氢氧化铝等多种矿物表面形成竞争吸附.

图1 有机质发生在无机胶体表面的部分吸附类型Fig.1 Adsorption of organic matter on inorganic colloid surfaces
目前,在胶体吸附能力的研究中,有机组分的作用尚未得到足够的重视,许多吸附研究仅以无机胶体作为主要的吸附体,因此并不能反映水环境中富含有机质的胶体的实际吸附性能;此外,以Langmuir为代表的吸附等温线模型无法从微观角度分析胶体的吸附机制以及有机组分对吸附机制产生的影响. 因此,在重视有机胶体作用的基础上,对胶体吸附机制的研究需要进一步细化,关注胶体形状、表面特性、配体性质变化对吸附能力的影响,并通过分子尺度的吸附模型进行模拟.
4.3 胶体的氧化还原反应
胶体在自然水体中的各类环境过程伴随着大量的氧化还原反应. 其中,如Fe、Al等胶体可由溶液中的低价态离子通过氧化作用形成,也能够在缺氧状态下通过还原反应重新溶解. 由于胶体常常活跃在沉积物-水界面等氧化条件变化剧烈的环境中,因此胶体能够频繁地参与到各种氧化还原过程之中.
胶体的氧化还原反应深刻影响着胶体的形态、胶体吸附物的价态,并最终影响胶体的迁移规律以及胶体结合污染物的环境效应. 例如,胶体中的水铁矿能促进其表面As(Ⅲ)氧化为As(Ⅴ),并能够在地下水等环境中通过微生物对铁水合物的还原反应释放溶解态的As(Ⅲ),从而根据环境的氧化还原性质控制As的赋存形态与毒性;在As和Cr共存的情况下,As(Ⅲ)与Cr(Ⅵ)能够在含铝铁水合物表面共同吸附,将毒性较高的As(Ⅲ)与Cr(Ⅵ)氧化还原为毒性较低的As(V)与Cr(Ⅲ),通过改变吸附离子的价态,降低重金属的毒性[109-110]. 硫作为一种对氧化还原反应敏感的元素,也能影响胶体及其结合物的氧化还原性质,在缺氧环境下,S(-Ⅱ)能够与水铁矿胶体形成FeS,并将一部分S(-Ⅱ)还原为S(0),从而降低水铁矿胶体的稳定性,加速胶体的团聚,因此,缺氧状态下,硫化物能够通过促进胶体溶解,加速剩余胶体团聚的方式大幅度降低胶体在孔隙介质中的含量以及迁移能力[111-112]. 以NOM为代表的有机胶体也具有活跃的氧化还原性质,有研究发现,NOM可以被微生物或地球化学还原剂还原,被还原的NOM在黑暗缺氧条件下可迅速将Cr(Ⅵ)还原为Cr(Ⅲ),形成稳定的HA-Cr(Ⅲ)胶体;而足够高浓度的HA则能够同时将Fe(Ⅲ)以及 Cr(Ⅵ)还原为Fe(Ⅱ)以及Cr(Ⅲ),并形成三者的复合胶体[113-114].
4.4 胶体的生物效应
由于胶体是多种污染物的重要载体,胶体及其结合物质的生物效应是胶体研究关注的重点. 胶体对各种水生动植物的生物利用度与毒性受到多种因素影响,包括胶体种类、组成、粒径与浓度、胶体与污染物的结合方式,以及胶体结合物与生物受体种类等.
Fe是浮游植物生成代谢、合成叶绿素以及细胞呼吸功能所必需的元素,尤其在海洋内,大量研究表明铁是限制浮游植物生产力的主要微量元素[115]. 胶体Fe作为水环境中Fe元素的重要组成部分,其生物利用度一直受到关注. 较大的尺寸与密度使得颗粒态Fe快速在水体中沉降,限制了生物对其的利用,而胶体Fe则能够像溶解Fe一样分布在水体之中,相关研究已经发现,胶体Fe可被硅藻等一系列浮游植物利用,但其生物利用度低于<1 kDa的真溶解态Fe. 在同样的条件下,根据浮游植物类型以及胶体来源的不同,胶体Fe的生物利用量被发现低于真溶解态Fe的6~31倍[116];同时,在胶体Fe内部,浮游植物对小粒径胶体的利用度高于大粒径胶体,因此,浮游植物对胶体Fe的利用可能基于胶体Fe自身的解离[117].
胶体也能够改变其结合物的生物效应. 在重金属中,胶体Cr的生物利用度在多个环境中均明显高于真溶解态Cr,例如,Pan等研究对比了不同来源的胶体态以及真溶解态Fe、Cr、Ag元素对绿贻贝和菲律宾蛤的生物有效性,发现胶体Fe、Cr、Ag均可被利用,且利用度远高于真溶解态的元素[118-119],而胶体Cd和Zn的生物利用率则在底栖动物中较低,在浮游生物中较高,显示出不同生物对胶体态金属元素利用度的差异[120], 其中胶体Zn的利用度被发现随着胶体有机碳含量的上升显著下降,表现出有机胶体对Zn生物活性的抑制效果[121]. 同时,不同来源胶体携带的金属在组织中的分布也各不相同,海岸胶体携带的金属更倾向于分布在消化腺组织,而河口胶体携带的金属倾向于分布在其他软组织中[120]. 除了重金属以外,也有少量研究关注了胶体中有机污染物的生态毒理效应,例如Sun等研究了胶体双氯芬酸在斑马鱼体内的生物积累效应,结果表明胶体的存在加速和增强了双氯芬酸在斑马鱼肌肉和脏器中的积累,且相对于高双氯芬酸浓度的环境,胶体在环境浓度下对双氯芬酸生物积累的增强效应更为显著[122]. 此外,Kang等则发现粒径在100 nm以下的天然胶体在环境浓度下能够引发斑马鱼的发育和表型畸形、氧化应激和代谢紊乱[123].
胶体及其结合物生物效应改变的原因目前尚不明确,部分实验初步探究了胶体中有机质含量、胶体来源地、胶体分子量以及胶体老化等因素的影响,但因为胶体组成的复杂性、生物物种的差异性等原因并未发现一致规律,而表征胶体性质以及胶体与污染物结合方式更增加了胶体结合影响污染物生物效应方面研究的难度.
5 胶体在水环境中的团聚与输移特性
5.1 胶体的团聚
团聚是胶体独特的动态特征. 胶体团聚后,胶体的运动将受重力作用逐渐沉降,同时,团聚也能改变胶体的尺寸,影响胶体在孔隙介质中的迁移能力,进而影响被胶体结合的污染物的迁移过程. 胶体团聚的过程受胶体粒子间的相互作用力控制,主要包括外界水动力影响以及与胶体自身体积、表面性质有关的吸引力和排斥力. 其中,最重要的是粒子间的范德华作用和双电层带来的引力与斥力. DLVO理论基于胶体粒子间这两种主要的相互作用以及胶体粒子间的距离,构建了预测胶体稳定性的模型[6].
DLVO理论下的胶体团聚行为包括两个反应过程:反应控制型(RLA)与扩散控制型(DLA),当胶体间存在排斥能垒时,胶体间的附着效率α<1,此时运动碰撞的胶体未必能形成聚集,团聚的速度由胶体间能否反应产生黏着决定,团聚速度较慢;当溶液电位变化导致胶体间的排斥能垒消失时,附着效率α=1,此时互相接触的颗粒都能形成聚集,从而达到胶体的最大团聚速率.
目前,胶体的团聚过程可以通过动态光散射(DLS)技术实时检测胶体颗粒的粒径与数量变化并通过聚集动力学模型来描述. 主要指标包括胶体的聚集速率以及相关影响因子的临界聚沉浓度(CCC). 通过观察水环境中胶体的团聚过程,在表面能垒降低的过程中,胶体的团聚速度逐渐上升,并最终稳定在最大团聚速度,这种由反应控制型团聚发展到扩散控制型团聚的过程证明了水环境中胶体的团聚符合DLVO理论[124].
对胶体团聚的研究表明,溶液的pH、离子强度、离子类型以及有机质的存在都能显著地影响胶体的稳定. pH值能够显著改变无机胶体的表面电势,通常,无机胶体在高pH值环境中拥有负电荷,在低pH环境中拥有正电荷,并在中间某pH值处电势为零,该pH值也被称为零电荷pH点(pHPZC). 显然,pH值越接近零电荷点,胶体间的静电斥力越小,越易于团聚;而pH值越远离零电荷点,斥力越大,越容易稳定. 对于有机胶体,有研究表明pH的上升会导致其分解成更小的分子量组分,而溶液离子强度上升则导致有机胶体的平均分子量升高[44]. 有机质通常表面携带负电荷,可以通过DLVO理论内的静电作用提高胶体的稳定性,但是在有机质浓度过高时,也会因桥联效应导致胶体聚集. 与携带负电的有机质相反,单价、二价阳离子都能有效降低胶体的表面负电荷,显著促进胶体的团聚[39].
5.2 胶体与污染物在地下水中的输移过程
与悬浮颗粒物相比,胶体在水体中不易于沉降,通过团聚形成大粒径颗粒后才能从水体中去除,而较小的粒径也使得胶体易于通过孔隙介质输送其携带的结合物质,因此,胶体在水环境中拥有独特的输移作用[125-127].
胶体能够有效促进多种重金属元素在土壤中的迁移[128-129],例如Baumann等在垃圾填埋场附近的地下水中发现了Fe、Cd、Co、Cu、Ni、Pb等一系列重金属与胶体的强烈结合以及共同迁移的现象[130]. 胶体的促进作用受胶体的产生、胶体与污染物的结合、胶体在介质中的运动3个过程的影响.
胶体具有多种产生机制,除了水动力条件变化导致固体颗粒分散破碎以外,所处环境的物理、化学变化也能产生相应的胶体. 例如,降水下渗、河流汇流时水体中离子强度下降,颗粒间的斥力增加,从而解聚形成胶体[131-132]. 胶体产生的同时将与介质中污染物结合,由于水环境中的胶体浓度通常较低,能被胶体显著促进输移的物质必须要具有易于与胶体结合的特点. 胶体在孔隙介质中的运动受到介质中的水体流动、介质的孔隙特征以及介质的过滤与吸附作用影响,在非饱和孔隙介质中,胶体的迁移额外受到液-气界面与表面张力的吸附作用,并有可能被固定在流体静置区域. 当胶体粒径大于介质的孔隙时,胶体将被固定在孔隙中;介质表面的吸附作用也能够将一部分活动胶体重新吸附[133-136]. 近年来,各类研究通过观测土柱出流实验中的穿透曲线(breakthrough curves, BTCs),积累了大量胶体迁移的观测数据与结果,并对胶体迁移中水流流速、胶体浓度、pH值、离子强度、有机质含量等一系列因素的影响进行了探究. 这些因素一方面可以通过DLVO理论内的作用关系影响胶体的团聚,进而影响胶体在孔隙中的运动;另一方面也能够影响污染物在胶体与其他介质之间的分配,并最终影响胶体与污染物的共同迁移. 例如Yang等通过室内模拟实验,发现水铝矿胶体在浓度较低(2.0×10-6mol/L)时对U(Ⅵ)的迁移具有促进作用,在浓度较高(5.0×10-5mol/L)时则因团聚起到阻碍作用,而土壤溶液中HA可以通过加强吸附、减少团聚的方式来促进胶体与污染物的迁移[137]. Chotpantarat等利用水饱和沙柱研究了不同pH值下蒙脱石胶体对Cd2+离子输移的影响,认为高pH值条件下Cd2+与胶体结合稳定,提高了胶体在介质中的迁移能力,反之低pH值条件则阻碍迁移[138].
在模型方面,胶体以及胶体促进迁移的数学模型基本原理通常基于胶体在多孔介质中运动的平流-扩散方程、胶体与固定多孔介质表面的关系以及污染物在液体、固体介质、胶体三态的分配,其中比较典型的有Corapcioglu根据平流-扩散-吸附方程提出的迁移模型,该模型中,胶体在介质内的传输作用取决于胶体溶液的布朗运动、浓度梯度下胶体的渗流以及该处胶体与溶液、固态之间的质量交换,并假定污染物在三态之间符合线性平衡吸附,最终构建胶体对污染物的迁移模拟. 近年来,多孔介质中胶体促进污染物迁移的模型已经开始将双位点附着、Langmuir型吸附、胶体的团聚纳入模型计算,以针对实际环境中可能出现的吸附位点阻塞、固相的非均质特性等实际情况.
此外,随着对环境内微生物作用的认知不断深入,许多研究者开始关注如病毒、细菌、原生生物等微生物在环境中的迁移行为,并使用“生物胶体”的概念来描述这些微生物在土壤中的迁移. 微生物污染物能够通过地下水传播疾病,对水资源安全构成很大的风险,因此,微生物在土壤的迁移是水环境研究中值得关注的现象. 与非生物胶体相似,微生物间也存在着黏着-分散运动,微生物间的黏着是一个复杂的过程,受到细菌和基质表面各种物理化学性质的影响,最显著的是其疏水性/亲水性和表面电荷[139]. 随后,微生物间形成的生物膜将进一步附着微生物形成体积更大的团聚体,从而影响生物胶体在迁移过程中的尺寸. 生物胶体在孔隙介质中也和非生物胶体一样在水动力、重力作用下迁移,受到孔隙介质的过滤、沉积吸附以及相关外部因素影响,例如,Wu等发现带负电荷蛋白质减少了细菌在石英砂中的沉积,促进了细菌的迁移,而正电荷胶体具有相反的作用,表明了静电作用对生物胶体迁移的控制[140]. 双酚A被发现能够利用其疏水性与细菌竞争石英砂表面沉积位点,从而促进细胞在石英砂介质中的迁移,且不受细胞类型、离子强度、离子价、细胞外聚合物质等因素的影响[141].
生物活性是生物胶体显著区别于非生物胶体的重要特征,微生物对环境中化学物质的浓度梯度产生趋向或离避响应的趋化性迁移是生物胶体在孔隙介质中运动的重要部分. 例如,Wang等发现,在天冬氨酸甲酯的诱导下,大肠杆菌在特定方向上的迁移总量相对于无诱导条件增长了18%[142]. 趋化性迁移对生物胶体的影响是复杂的,受到微生物的种类,相关化学物质的类型、分布以及微生物的趋化能力影响,因此趋化性迁移既可能促进,也能够抑制生物胶体的迁移,例如,垂直分布的萘使得恶臭假单胞菌G7的纵向分散增加了3倍,水平流动的回收率降低了43%,极大地增加了恶臭假单胞菌G7在介质中的停留时间[143].
5.3 胶体与污染物在地表水中的输移过程
在地表水体中,稳定的胶体通常在水流作用下伴随移动,与溶解态物质相比,胶体输移的特点在于其动态的粒径能够引发自身的形态变化,并进一步影响污染物的迁移. 如图2所示,“胶体泵”便是用于描述胶体在地表水中吸附、团聚过程的一种理论[13].
“胶体泵”理论由Honeyman和Santschi于1989年提出,该理论提出了一种概念和数学模型,认为溶解态金属能够通过胶体粒子形成易于沉降的颗粒物;该过程包括了金属在胶体表面的快速吸附以及胶体与颗粒物的缓慢聚集两个速率不同的步骤.
Honeyman根据颗粒物团聚模型构建了以颗粒浓度Cp为变量的团聚方程:
(6)
式中,Cp为水体中的颗粒物浓度,mg/L;Bds、Bsh和Bb分别是沉降(差向絮凝)、流动剪切力(同向絮凝)和布朗运动(异向絮凝)导致颗粒物聚集的速率系数,d-1.Bds、Bsh和Bb具体数值可根据环境条件由相应公式计算,该方程可以简化为以B′b为总聚集速率系数的形式:
(7)
结合重金属在胶体中的摩尔浓度系数Гc(mol/g),可将颗粒物以及其中的重金属含量与胶体的动态聚集联系起来:
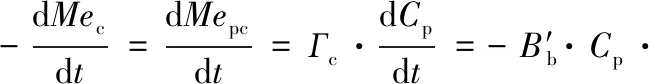
(8)
式中,Mec、Mepc为重金属在胶体以及颗粒物中的浓度. 在此基础上,根据现场和模拟实验的数据,研究发现在深海环境、透光带、沿海和河口系统中,胶体泵模型均具有一定的适用性. 1996年,为了验证“胶体泵”理论,Stordal等使用203Hg作为示踪剂,追踪到了胶体快速吸附203Hg后逐渐形成大于0.45 μm颗粒物的过程. 与“胶体泵”模型对比,Stordal采用不可逆的一阶反应模型对胶体团聚过程进行了拟合,发现实验中的团聚速率与“胶体泵”计算结果大体相符,并证明布朗运动是胶体发生团聚最主要的动力[144].
胶体泵的作用也在实地研究中得到了印证,Kimball调查了阿肯色河不同河段中重金属在溶解态、胶体态、颗粒态三态之间的浓度,发现在矿水排入的情况下,胶体在下游50 km处即通过团聚沉降的方式减少了一半的胶体态重金属,而溶解态重金属的浓度则几乎不发生变化,表明了胶体能够将重金属固定在沉积物中,降低部分重金属在河道中的扩散[145]. Stople、Zhou、Ren等也在河流交汇、河口混合带中观测到了由于水化学环境变化而导致的胶体团聚、固定重金属的现象[50,74,146].
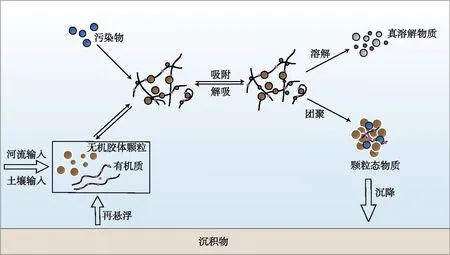
图2 天然水体中胶体对污染物的迁移转化作用Fig.2 Effect of colloid on migration and transformation of pollutants in water environment
此外,胶体在河湖水体中特有的再悬浮过程中的作用也在部分研究中得到了重视. 在风浪或水流扰动导致再悬浮的过程中,沉积物中的营养盐、重金属等污染物重新暴露到上覆水中,化学环境的变化以及水力破碎作用都有助于形成胶体并影响污染物的释放[147]. 例如,孙小静等发现在风浪扰动下,太湖水体中的胶体态氮、磷呈上升趋势,在扰动结束后,胶体氮、磷占据总氮、磷的比例显著上升,表明沉积物释放的胶体态氮、磷在水体中拥有更长的停留时间,增加了水体中营养盐的浓度[148-149]. 张战平等则观察到了风浪引起重金属释放,其中胶体态的Mn、Cu和Pb含量与比例在风浪作用下显著上升,而胶体态Zn则体现为去除效应[150].
6 总结与展望
胶体概念的引入,突出了水体中性质活跃且受重力影响微弱的亚微米级物质的作用,为研究微量物质的环境行为提供了基础. 胶体的研究有助于更准确地认识不同形态污染物在环境中的反应活性,并从微观层面阐述相关机制,进而有利于理解污染物对环境质量的影响,控制修复其造成的环境危害.
当前,水环境中胶体的研究初步实现了胶体的提取、表征,研究了水环境中胶体的形态、组成以及其中污染物的含量,并对胶体的吸附、团聚等基础反应进行了观测和机制探究. 研究证实了胶体是污染物在环境中重要的赋存形式与迁移载体,并总结了胶体在部分环境过程中的作用机理与生物效应. 尽管胶体的研究已经日益深入,其中依然存在着不少问题与挑战:
1)当前对胶体结合污染物的能力主要关注胶体的吸附能力,对胶体生成过程中以再沉淀形式结合污染物的机制研究较少. 对胶体吸附机制的研究大多只针对单一的有机或无机胶体展开,主要关注了pH值、离子强度等外界因素对吸附能力的影响,对有机、无机胶体混合体系的研究较少;吸附实验中对胶体可能的团聚、光降解等过程导致的粒径、成分变化不够重视,主要集中于稳定、静态的胶体. 此外,当前应用的经验吸附模型也不能从分子层面解释胶体吸附机制以及吸附能力的变化. 未来应加强对胶体吸附行为的精确化、机理化研究,构建能够涵盖胶体有机组分与无机组分相互作用的胶体-污染物吸附模型.
2)现有研究主要集中在胶体对各类环境物质的分布、输移规律的影响,对胶体吸附结合后是否造成污染物性质转变,胶体态物质是否拥有特殊的生物活性以及毒理作用的研究较为缺乏. 胶体具有复杂的生物效应,受到胶体自身性质、结合污染物类型、胶体与污染物的结合形式以及不同生物种类的影响,当前研究仅探究了少量胶体污染物的生物利用度,对其背后的毒理机制、各因素影响尚缺乏了解. 其次,胶体的动态变化使得胶体态物质无法脱离溶解态、颗粒态物质单独存在,胶体物质也可能随时向溶解态、颗粒物转化,在研究胶体物质的性质与作用时,如何排除颗粒态、溶解态物质的干扰作用依然有待进一步研究.
3)河湖水体中存在着复杂的环境变化,如水动力变化、生物作用以及光化学反应,这些环境过程深刻地影响着水体中的理化性质以及相关物质的迁移转化规律. 对于胶体而言,在河湖水体复杂的环境过程中如何变化、如何与外界环境相互作用尚未得到充分研究. 虽然已有部分研究在再悬浮、河流混合等过程中对胶体态物质进行了观测,但主要集中在胶体内污染物的含量变化上,对这些过程在改变水体的理化指标的同时如何影响着胶体的数量、组成、形态,进而影响胶体的吸附、团聚特性以及胶体结合的污染物性质转变缺乏详细数据与机制理解,应加强胶体室内模拟研究与野外实际调研的联系,进一步揭示胶体在各类环境过程中的作用与意义.
4)环境中胶体的概念不断推广,应重视新的胶体类型与来源. 包括细菌、病毒、藻类等微生物因为具有符合胶体的粒径以及在孔隙介质中与胶体具有相似的输移行为,因而被部分研究称为“生物胶体”. 从胶体的角度更加易于理解阐述部分微生物的环境行为,表明了胶体的概念不再局限于强调胶体与溶解态、颗粒态物质的区别,而是逐渐拓展到研究一切微观物质的表面性质、活动能力以及最终的环境效应上. 此外,目前的胶体研究仅限于水圈与岩石圈中,而大气中的气溶胶显然具有符合胶体定义的粒径. 气溶胶沉降是否是水环境中胶体的重要来源,水环境中胶体有无可能扩散到大气之中,以及胶体在水体与大气中迁移的环境意义还缺乏相应研究.
猜你喜欢
石油沥青(2022年2期)2022-05-23 13:02:36
建材发展导向(2022年2期)2022-03-08 01:43:38
中国食用菌(2021年10期)2021-11-04 06:23:20
建材发展导向(2021年14期)2021-08-23 00:57:44
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
北京航空航天大学学报(2017年2期)2017-11-24 05:24:51
中国环境监察(2017年3期)2017-05-14 06:19:02
天津科技大学学报(2015年4期)2015-04-16 04:55:09
中国卫生标准管理(2015年14期)2015-01-27 02:24:27
西南医科大学学报(2014年6期)2014-03-20 15:43:50
