
醇羟基对碳酸钙矿物成核物相及界面能影响研究
2021-01-11 07:08王洪涛江锐捷
高校地质学报 2020年6期
杨 阳,朱 辰,王洪涛,花 浩,江锐捷,毛 玮,赵 良*
1.南京大学 地球科学与工程学院,南京 210023;2.南京大学 地理与海洋科学学院,南京 210023
1 研究背景
碳酸钙矿物是碳元素的重要地质储库之一(Berner et al.,1983; Berner, 2003),具有三种常见的无水形式同质异象体:方解石、文石和球文石。方解石是地质历史记录中最常见的碳酸钙矿物,常温常压下热力学最稳定。其中生物成因方解石包含了丰富的地球化学信息(Mann, 1983),特别是有孔虫方解石骨骼保存的元素、同位素信息可用于古气候、古海洋等古环境重建研究(Elderfield and Ganssen, 2000)。文石相比方解石不稳定,在成岩作用中转变为方解石(Budd, 1988),但文石是现代壳类动物壳体常见的碳酸钙矿物相(Falini et al., 1996)。球文石最不稳定(Lippmann, 2012),但与生物作用紧密相关,已在活的有孔虫壳体(Jacob et al., 2017),含细菌土壤(Rodriguez-Navarro et al., 2007; Lian et al., 2006), 蛋 壳(Board and Perrott, 1979; Portugal et al., 2017),珍珠(Soldati et al., 2008; Addadi et al., 2006; Gauldie et al., 1997), 鱼耳石(Campana, 1999; Carlström, 1963),海鞘骨针(Kabalah-Amitai et al., 2013; Lowenstam et al.,1975),动物尿结石(Neumann et al., 1994; Mair and Osborn., 1986),人肾结石(Sutor and Wooley,1968; Bills and Lewis, 1975),心脏瓣膜(Kabalah-Amitai et al., 2013),甚至植物(Wightman et al.,2018)中被发现,然而针对活体有孔虫(Jacob et al., 2017)、蜗牛(Hasse et al., 2000)、蚌类 (Spann et al., 2010) 的研究发现,球文石极有可能先与方解石或文石出现,作为前驱体参与骨骼形成。
已有研究表明,碳酸钙物相控制因素复杂,温度(Ogino et al., 1987),过饱和度(Spanos et al.,1998),无机物(Morse et al., 1997),有机物(Jimoh et al., 2018)等都会影响形成的最终物相。生物是通过何种机制控制碳酸钙物相尚不清楚。有研究者认为生物体中的多糖有机质可通过改变其表面电荷或结构,从而控制溶液和碳酸钙物相的界面能,进而控制生物成矿过程(Giuffre et al., 2013);也有研究者认为生物体富含的蛋白质通过羧化氨基酸获得带负电位点,从而与Ca2+结合控制碳酸钙的形成(Weiner et al., 1975);亦或是通过囊泡包裹并稳定化亚稳定相(比如无定形碳酸钙)物质再形成更稳定的碳酸钙物相(Mass et al., 2017)。这些机理均要求量化描述矿物界面的对溶液物质的响应,而界面能反映了矿物从溶液中沉淀的难易程度(Mullin, 2001),界面能越高,矿物沉淀所需要克服的能量就越高,越不利于矿物的形成。矿物结构、吸附作用、表面电荷均会影响界面能的大小。生物体主要通过何种因素控制界面能,进而控制碳酸钙矿物物相,是十分值得研究的问题。
羟基是生物体系中常见的基团,广泛存在于醇类,多糖类,氨基酸、蛋白质等物质中,已有研究表明,含有羟基的多糖类物质可以作为碳酸钙的成核位点(Giuffre et al., 2013)。此外,与单羟基小分子醇相比,多羟基醇可有效促进球文石亚稳定像的形成(Trushina et al., 2014)。然而,由于缺乏在醇分子参与下碳酸钙矿物溶解度积数据,针对碳酸钙物相在醇类有机物质参与下的界面能研究较少(Manoli et al., 2000a)。针对醇类物质羟基对界面能的影响以及控制机制缺乏全面定量化的研究。
本研究试图通过标定碳酸钙在多羟基醇—水混合溶剂体系下的溶解度积,定量化研究乙醇、乙二醇、丙三醇对碳酸钙矿物物相以及界面能的影响,探究羟基数量对碳酸钙成核过程的控制机理,以便更好地理解碳酸钙物质的生物成矿过程及其地球化学行为。
2 实验方法
2.1 成核试验
实验中所使用的水为去离子水,其电阻率大于18.2 MΩ·cm-1,使用的乙醇、乙二醇、丙三醇等醇溶剂为分析纯,分析纯NaHCO3与CaCl2固体分别溶解于去离子水中,配置成0.1 M 的母液并通过0.22 μm滤膜过滤去除可能存在的固体杂质。实验所需的各种浓度的NaHCO3及CaCl2都由以上两种溶液稀释得到并定期配置,以减小反复配置低浓度溶液带来的误差。所用仪器包括:磁力搅拌仪(旋涡B11-2 型恒温磁力搅拌仪,上海司乐仪器有限公司)、台式水质分析仪(雷磁DZS-708 多参数水质分析仪,上海雷磁有限公司)以及低温恒温槽(DC-0506,上海舜宇恒平科学仪器有限公司)。反应装置如图1所示。
具体实验步骤如下:

图1 成核实验装置图Fig.1 Scheme of devices for nucleation experiment
(1) 取特定量的0.1 M的NaHCO3母液与去离子水以及特定体积醇(乙醇、乙二醇或丙三醇)进行混合(混合物1);
(2)取特定量的0.1 M的CaCl2母液与去离子水进行混合(混合物2)。溶液密封放在恒温水浴箱内,保证温度为25℃;
(3)混合物1和混合物2的数学体积之和为100 mL,实际体积通过实验校正获得,具体实验条件见表1;
(4)把混合物1移到夹套烧杯水浴循环,温度设置为25℃,磁力搅拌仪转速300 rpm。同时检测体系pH、电导率,数据记录时间间隔为1 s。待系统温度稳定后,向其中迅速加入温度相同的混合物2,实现Ca2+与CO32-的接触混合。使用这种混合方式可以将混合时间降至1 s,以此来降低混合过程对诱导时间测量的影响;
(5)待溶液浑浊且pH、电导率等数据均稳定后,把实验反应所得的固体过滤并用去离子水洗涤,置于常温下的干燥皿中。实验停止后,用盐酸清洗夹套烧杯、pH、电导率以及温度探头,以防有剩余的晶体作为晶核影响下一组实验,导致新实验产生异相成核;
(6)干燥清洁后的固体,采用X射线衍射分析(XRD)物相。分析仪采用铜靶,2θ角度为3°~60°(Bruker D2 Phaser)。获得数据后,采用Jade软件进行分析,确定反应得到固体的物相种类。
2.2 碳酸钙溶解度实验
使用分析纯CaCO3作为标样(南京化学试剂股份有限公司),XRD分析表明为纯的方解石物相。球文石采用文献中的方法合成(Kralj et al., 1990),XRD 分析确认纯的球文石物相。取大于10 mg的方解石或者球文石固体置入离心管中,分别装入约50 mL 特定体积比的醇 —水混合溶液中并混合均匀。将离心管置于25℃恒温摇床中72 小时后,以5000 rpm 离心10 min,取上层清液使用电感耦合等离子体发射光谱仪(ICP-OES,Thermo Scientific)进行Ca 浓度测量。同时测量清液的pH 值。
3 数据与结果
3.1 碳酸钙物相
XRD结果表明(图2),在25℃温度条件下,当NaHCO3与CaCl2在100 mL 的纯水体系下反应时,生成的固体产物都为单一的方解石物相,没有其它的碳酸钙同质异象固体产物生成。此外,当在体系中加入5 mL或10 mL乙醇且溶液总体积控制为100 mL时,反应生成的固体产物仍然为单一的方解石物相。然而,将单羟基的乙醇替换为5 mL 或10 mL 的双羟基的乙二醇后,反应生成的固体产物为球文石和方解石的混合物。根据Kontoyannis等(2000)提出的XRD碳酸钙物相定量方法,估算乙二醇实验中沉淀的固相中球文石占65%,方解石占35%。将乙二醇替换为5 mL或10 mL 含有三羟基的丙三醇后,反应生成的固体产物转变为单一的球文石物相。

图2 成核实验固体产物XRD 谱图Fig.2 XRD pattern of solid products from nucleation experiments
3.2 碳酸钙矿物成核诱导时间
对于典型的碳酸钙结晶过程来说,诱导时间就是指从溶液建立过饱和到观察到碳酸钙形成之间的时间间隔。最先从溶液中形成的纳米碳酸钙颗粒称为晶核,而晶核需要长大到足够大以后才能被仪器检测到,实测的诱导时间可以用下式表达(Mullin, 2001):

其中tr表示系统中原子簇达到准稳态分布所需时间,tn表示形成稳定晶核所需的时间,tg表示晶核生长到可检测大小所需的时间。tr的大小主要与系统的粘度和扩散性有关,在溶液中tr的数量级一般在10-8s左右(Mullin, 2001),与实际测得的碳酸钙的成核时间相比(11 s~22 min)可以忽略不计。tg这一时间的长短与所使用检测手段的灵敏度有关,检测手段灵敏度越高,也就意味着tg就越短,实际测得的诱导时间就会越接近于成核时间。对于本次研究所用的电导率检测来说,可实现1 μS/cm 的灵敏度,对应于约7 nM的KCl浓度或者0.35 nM的CaCO3沉淀量变化。这样的灵敏度可以满足实验需求。
在NaHCO3与CaCl2溶液混合后,溶液的电导率会出现一个突跃,之后形成一个较为稳定的平台,一定时间后电导率下降,肉眼可见溶液变浑浊。将溶液突变的时间点作为过饱和建立的开始,电导率开始下降的时间点确定为碳酸钙被检测到的时间,两点的时间差为实验获得的诱导时间。具体的数据处理方法见Zhao等(2013)。
表1列出了不同NaHCO3与CaCl2浓度,不同醇类分子及体积含量下,通过对混合后CaCO3过饱和溶液电导率进行监测获得的诱导时间数据。实验数据表明在相同溶剂环境下,CaCO3物相的成核诱导时间基本与溶液中NaHCO3与CaCl2浓度成反比,浓度越高诱导时间越短。在NaHCO3和CaCl2浓度相近的情况下,加入的醇的种类可显著影响碳酸钙矿物的成核诱导时间。比如,在低浓度下(~0.005 M CaCl2和NaHCO3),不加入醇类分子的条件下的碳酸钙成核诱导时间为85 s,而分别加入5 mL乙醇、乙二醇和丙三醇后,成核诱导时间逐渐增加为110 s、123 s 和1374 s。当将醇类组分由5 mL增加至10 mL时,诱导时间会有一定程度增加。当溶液CaCl2和NaHCO3浓度增加至0.015 M时,含有5 mL丙三醇体系的诱导时间(131 s)仍然高于水体系下的诱导时间(65 s),而其它醇类的诱导时间(5 mL乙醇26 s、5 mL乙二醇15 s、10 mL乙二醇18 s)低于水体系下的诱导时间。由此可见,诱导时间对于醇类的种类和浓度具有不同的响应。图3展示了诱导时间在不同醇类及浓度条件下随溶液Ca2+和CO32-活度乘积的变化。总体而言碳酸钙诱导时间随活度积的增加而呈现指数下降,醇类的加入显著影响诱导时间的绝对值。
3.3 碳酸钙物相在不同醇—水混合溶剂中的溶解度
表2列出了25℃ 温度下,球文石和方解石物相在不同比例的醇—水混合溶剂体系中的溶解度积常数(Ksp)。水溶剂条件下实验获得的方解石和球文石的溶解度积常数(Ksp)为10-8.37和10-7.94这与文献中的数值10-8.48和10-7.91基本一致(Plummer and Busenberg, 1982)。醇的加入会显著降低碳酸钙物相的溶解度,其中方解石和球文石的Ksp在丙三醇水体系中呈现比其它体系更低的数值。此外,碳酸钙物相的Ksp在同种醇—水体系中随着醇类的体积百分比的增加而进一步降低。为了验证实验获得的实验所得的Ksp的正确性,可将其与文献的值做比较。以乙醇为例,28℃时10%体积分数的乙醇—水混合物中方解石Ksp为10-8.70(Gomma,2012),因温度的原因略高于实验值10-9.02。此外,文献中25℃条件下获得的10%乙二醇—水溶液中方解石Ksp在10-8.65和10-9.19之间,而本文实验获得的值为10-9.01,与其它研究者结果相吻合。因此,本文中使用的实验方法可以有效地获得方解石和球文石在醇 —水混合体系下的Ksp,进而可以计算醇 —水体系下的实际过饱和度(Ω)。
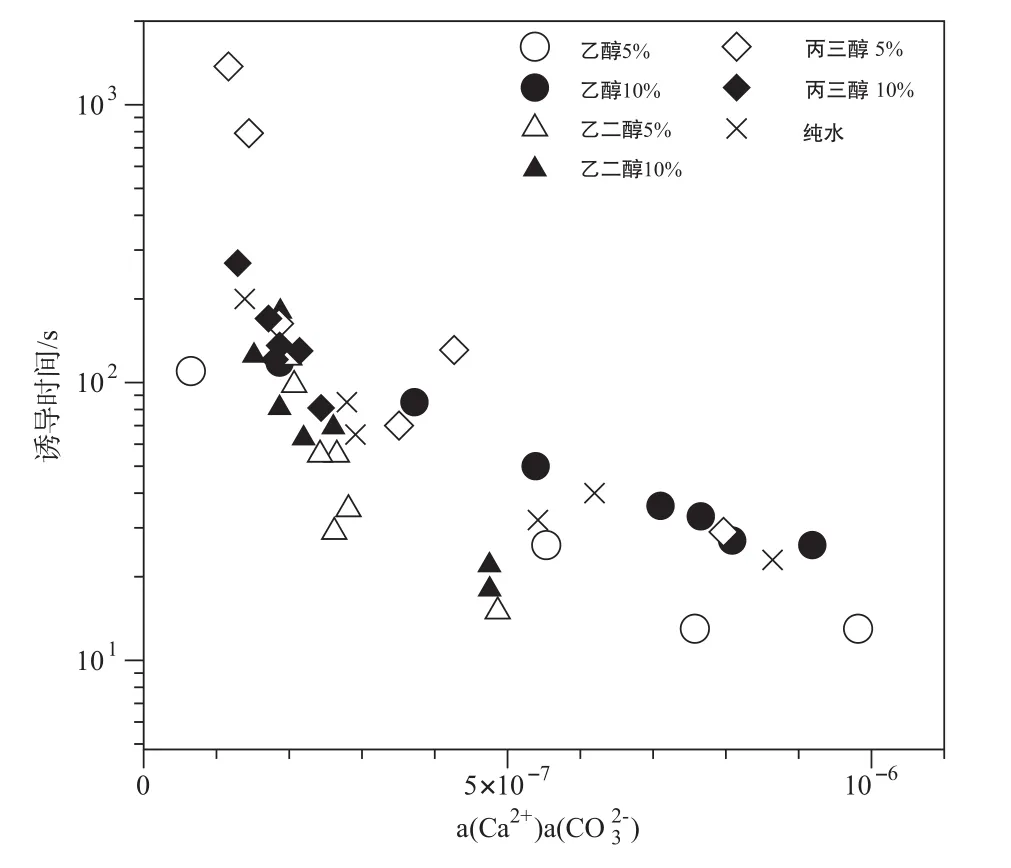
图3 不同离子活度积条件下诱导时间随醇—水溶剂性质的变化Fig.3 Variation of induction time with alcohol-water mixtures under different ionic activity products

表1 成核诱导时间实验条件(Ω为相对实验获得纯物相的过饱和度)Table 1 Experimental conditions of nucleation experiments (Ω represents the supersaturation degree relative to the pure phase)

表2 25℃ 下方解石和球文石在不同醇—水体系下的溶解度Table 2 Solubility of calcite and aragonite in alcohol-water mixtures under 25℃
4 讨论与分析
4.1 醇类物质对碳酸钙成核界面能的影响
矿物—溶液界面的界面能是评价成核过程的重要参数,它代表了矿物从过饱和溶液中形成所要克服的能垒高低。成核时系统吉布斯自由能变化主要是由体积自由能的降低和界面自由能的增加共同决定的。Ca2+和CO2-3离子从溶液态转变为固态团簇会导致体积自由能降低,另一方面,新形成的固液界面具有额外的能量(即界面能)使得体系总界面能增加。当CaCO3固体团簇半径极小时,界面能发挥决定性作用,使的系统吉布斯自由能增加。而随着团簇粒径的增加,体积自由能的降低使得系统吉布斯自由能的上升幅度越来越慢,当团簇达到临界尺寸时,系统吉布斯自由能达到最大值,之后系统吉布斯自由能持续降低,此后团簇快速地生长,完成成核过程(闵乃本,1982; Mullin, 2001)。这一过程称为经典成核理论(Classical nucleation theory, CNT),该过程的数学表示如式2所示。

其中ΔG(n)代表体系的吉布斯自由能变化,-nΔµ代表固体自由能的下降,aγn2/3代表整体界面能的增加。n为团簇中分子的数目,Δµ为碳酸钙分子在溶液中与在固相中的化学式差值,a为形状因子,γ为界面能。当时为体系ΔG(n)最高点,通过求解该微分方程可获得该点的团簇临界尺寸n*以及对应的ΔG(n*)。若成核速率与ΔG(n*)符合阿勒尼乌斯关系式且诱导时间与成核速率呈反比,则可获得诱导时间的定量描述(Zhu et al., 2016a)如下式:

其中tind为诱导时间,kB为玻尔兹曼常数,T为开尔文温度,Ω为过饱和度,B为常数。Ω的定义如下:

由于本研究所用的醇类物质电离常数(pKa)较低,比如25℃时乙醇为10-15.5,乙二醇为10-14.22,丙三醇为10-14.15,均与水的电离常数相近10-14。因此,计算离子活度积时假设醇类物质提供质子的能力与水类似。此外,Lu等(2010)研究了乙二醇—水体系下和离子活度系数与乙二醇含量之间的关系。结果表明,当温度为25℃,离子强度为0.1 M时,10%体积分数下和离子活度系数分别为0.84和0.97,相比于水体系下分别下降了16%和3%。因此,由于加入的醇体积分数均小于10%,因此,计算时也假设各离子的活度系数近似等于水体系下的活度系数。在此基础上,使用Phreeqc软件计算混合溶液中和的活度积。再结合实验获得的各醇 —水体系下方解石和球文石的溶解度积,便可计算出实际的过饱和度。
图4(a)展示了不同过饱和度情况下,醇类分子对诱导时间的影响。由于乙二醇条件下为球文石和方解石的混合物,因此没有计算过饱和度。可以看出,在相同过饱和度条件下,醇类物质的加入会增加诱导时间,降低碳酸钙矿物的成核速率。在低过饱和度范围内(Ω<200),乙二醇—水体系内的诱导时间要高于乙醇—水体系和水体系,而在高过饱和度范围内(Ω>500),乙醇—水体系内的诱导时间要高于水、丙三醇—水体系。此外,对于相同的醇—水体系,增加醇的体积分数会增加相同过饱和度下的诱导时间。根据式(4)将过饱和度和诱导时间进行变换后如图4(b),可知(lnΩ)-2- l n(tind)呈线性关系,符合经典成核理论。通过拟合的斜率可以计算出醇—水体系下的碳酸钙矿物界面能。结果显示,本研究获得的水体系下的方解石界面能为36.5 mJ/m2,与文献中37.8 mJ/m2( Westin and Rasmuson, 2005)的数值相近。加入5%和10%的乙醇后,方解石界面能逐渐增加至39.9和48.9 mJ/m2。与之类似,文献中水体系下球文石的界面能为40.7 mJ/m2(Gómez-Morales et al., 1996a)或37.3( Verdoes et al.,1992),加入5%和10%的丙三醇后球文石的界面能逐渐增加至47.0和53.5 mJ/m2。文献中25℃条件下10%体积分数的乙醇、2-丙醇以及二甘醇—水体系中球文石界面能分别为414544 mJ/m2(Manoli et al., 2000a)。乙醇体系下的球文石界面能同样高于方解石,这与水体系下的规律相一致。同样体积分数下,丙三醇—水体系下球文石界面能高于乙醇—水体系下方解石的界面能。
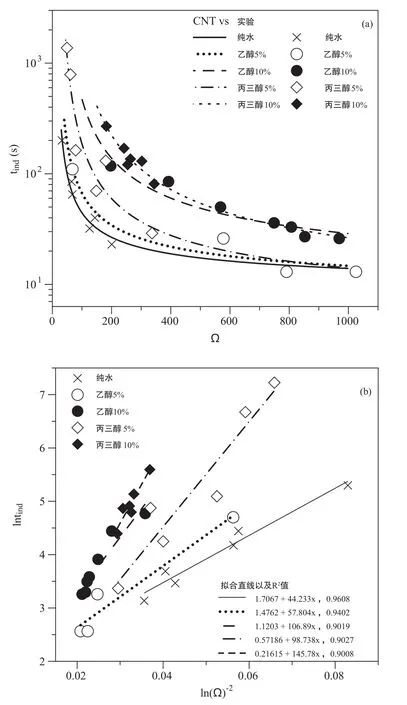
图4 采用经典成核理论分析诱导时间数据Fig.4 The induction times are analyzed using classical nucleation theory
为了经一步理解醇类物质对碳酸钙矿物界面能的影响机理,还需从最基本的分子层面来讨论。对于真空中的矿物表面来讲,由于表面断键的存在,位于矿物表面的原子具有比晶体内部原子更高的能量,此时的界面能主要取决于表面化学键的密度和能量。当矿物从真空进入水溶液中后表面原子在溶剂分子的作用下发生溶剂化作用、质子化作用,断键的能量被一定程度上释放,界面能有相当程度的降低。若溶液中存在可以与矿物表面发生吸附作用的分子或离子,那么与表面原子间的络合作用会进一步降低界面的能量。进一步的,通常矿物表面是带有电荷的,那么矿物表面与溶液间便会形成电场,而电场中表面带电原子还具有电势能,当溶液中的异号离子受矿物表面电荷吸而靠近时会在表面形成双电层,从而使电场更快的衰减,降低矿物表面原子的电势能(Parks, 1984; Zhu et al., 2016a)。因此,对于溶液中的矿物表面,其界面能受矿物结构、表面吸附以及表面电势能三者共同决定。他们之间的关系可用下式描述:

其中γ0表示矿物结构决定的界面能,R为理想气体常数,Γ为吸附物种,σ为电荷,ψ为电荷对应的电势,ψ0代表表面电势。
首先讨论醇类物质的介电常数对碳酸钙界面能中电势能部分的影响。对于一个球形的颗粒而言,其表面电荷σ0正比于溶液介电常数D和表面电势ψ0的乘积。因此,如果醇类物质加入如果会降低溶液的介电常数,则会降低碳酸钙表面原子的电势能。根据式(5),碳酸钙颗粒的界面能则会上升。已知25℃时纯水、5%和10%体积分数乙醇—水混合物的介电常数分别为78.5,76.1和74.0,而5%和10%体积分数丙三醇 — 水混合物的介电常数为76.6和75。由此可见,相同体积分数条件下,乙醇会更显著的减小混合溶液介电常数。此外,由于方解石密度高于球文石,可推测相同pH条件下,方解石界面的电荷密度应高于球文石。所以,理论上,介电常数的变化对方解石的影响应该更大。然而,将醇体积分数与界面能进行线性拟合可发现(图5),球文石是在丙三醇—水溶液中的界面能变化率(1.4±0.2 mJ-1/m-2)高于方解石在乙醇—水溶液中的变化率(1.1±0.2 mJ-1/m-2)。与设想不一致。说明介电常数的变化并不是决定碳酸钙在醇—水体系下界面能的主要原因。

图5 碳酸盐矿物界面能随有机—水溶剂比例变化趋势Fig.5 Variation of CaCO3 interfacial energy with alcohol-water ratios
除了溶剂性质以外,另一个决定界面能高低的因素为表面电荷的多少。乙醇分子具有一个羟基(-OH)和一个甲基(-CH3),这一结构使其同时具有亲水性和疏水性,具有羟基的一端靠近固体表面,甲基端朝外不参与质子化过程(Cooke et al., 2010)。因此,乙醇的在碳酸钙矿物表面的吸附会降低界面的电荷密度,进一步增加碳酸钙的界面能。另一方面,丙三醇具有三个羟基,一个羟基参与表面吸附作用,其它羟基仍可以参与质子化过程,理论上丙三醇吸附对表面电荷密度的影响应低于乙醇,对界面能的影响应该低于乙醇。这仍然与实验结果相悖。
决定界面能大小的另外一个重要因素为吸附位的多少。若醇类物质与碳酸钙表面吸附位为一对一的相互作用,醇分子吸附后可能会存在“位阻”效应,阻碍相邻吸附位的吸附过程,变相降低了吸附位密度,从而增加界面能。理论上吸附分子的体积越大,吸附位密度越高,那么空间位阻效应应该越明显。假设吸附位密度与矿物单分子体积成反比,那么方解石、球文石的单分子体积分别为6.13×10-29和6.27×10-29m3,那么方解石表面吸附位应该更密集。另一方面,乙醇分子小于丙三醇。二者结合考虑吸附作用对界面能的影响应该相近,这与实验结果相吻合。此外,三水菱镁矿界面能在不同DMF体积分数下的研究表明(Zhao et al., 2013),由于三水菱镁矿分子体积为1.24×10-28m3,远大于碳酸钙,因此其受有机分子吸附的影响应该小与碳酸钙体系。文献中(Zhao et al., 2013)三水菱镁矿界面能随DMF体积百分比的变化率为0.45±0.02 mJ-1/m-2,远小于方解石的1.1±0.02 mJ-1/m-2和球文石的1.4±0.02 mJ-1/m-2。因此,可认为,醇类分子对碳酸钙矿物界面能的影响主要通过空间占位的位阻效应完成,吸附位密度越大,醇类分子体积越大,吸附位密度降低越明显,界面能越高。进一步将10%体积分数的二甘醇—水体系下球文石界面能44 mJ/m2与丙三醇体系下的53.5 mJ/m2作比较,尽管二甘醇和丙三醇的分子量相近,但由于丙三醇的羟基数量更多,形成氢键的能力更强,丙三醇可能形成分子团簇,进一步放大位阻效应,形成了更高的球文石界面能。
经典成核理论(式2)的截距反映了无穷大过饱和情况下分子向团簇聚合的速率。纯水体系下的截距为1.71 高于5% 和10% 的乙醇—水体系下的1.47 和1.12 以及5% 和10% 的丙三醇—水体系下的0.57 和0.22。这一结果表明,相比于纯水体系,醇的加入一定程度增加了碳酸钙矿物表面与离子的聚合速率。这与醇—水体系下较高的方解石界面能相对应,因为较高的界面能意味着较高的表面分子反应活性。针对乙醇分子在方解石表面的实验和分子模拟研究认为,醇的羟基与碳酸钙矿物表面结合从而将表面水分子置换羟基吸附使得甲基朝向晶体表面外侧,而甲基为疏水的惰性基团(Cooke et al.,2010; Sand et al., 2010)。根据相似相容原理,甲基的存在会提升矿物颗粒在水中的界面能但有利于与其它具有甲基表面结构的团簇聚合。
4.2 醇类物质对碳酸钙物相的控制机理
已有的针对碳酸钙物相的研究发现,多种无机物或者有机物都可以促进球文石的形成(Trushina et al., 2014)。比如氨分子、铵根离子、硝酸等无机物,以及氨基酸、蛋白质和蔗糖等有机质。除此以外,聚合物常表现出对球文石的高选择性,比如聚苯乙烯磺酸盐,聚丙烯酸,聚乙烯吡咯烷酮,聚乙烯醇,多元羧酸,羧甲基菊粉,树枝状大分子,杯芳烃等。特别的,醇类物质对碳酸钙物相的控制作用已经被发现。例如,在25℃下10% 体积分数的乙醇、2-丙醇以及二甘醇—水体系中,可以形成纯的球文石物相(Manoli et al., 2000a);纯乙二醇,1,2-丙二醇和甘油溶剂条件下,通过高温加热和形成纯的球文石(Li et al., 2002);而24℃下10% 和50% 乙醇、1-丙醇和2-丙醇可以在先形成无定形碳酸钙的条件下逐渐沉淀球文石等混合物相(Sand et al.,2012);25℃ 条件下乙二醇的加入会提升球文石在产物中与方解石的比例(Flaten et al., 2010)。总体而言,高浓度的有机物浓度、高分子量的聚合物以及高基团数量的树枝状大分子都有利于球文石的形成。Hu等(2012) 认为,球文石的形成是通过纳米颗粒的聚合来实现的,而有机物与球文石表面的吸附作用决定了颗粒的稳定程度。对于本研究中乙醇-水体系而言,乙醇的球文石的稳定化作用较弱,导致在过饱和度下降后球文石纳米颗粒溶解,不利于球文石的形成。随着乙二醇和丙三醇分子中羟基数量的增加,与球文石表面结合越强,也就有利于球文石的稳定,不容易进行再溶解。这一机理被用来解释双亲水嵌段共聚物对球文石的选择性(Yu et al., 2003)。但这一解释与本研究的界面能结论有冲突,因为无论是方解石还是球文石,其界面能都随着醇类物质含量的上升而增加,并不支持醇类物质使得颗粒更稳定这一设想。还有理论认为羟基的吸附在球文石晶核表面使得其界面能比其它两种物相(方解石、文石)要低,使得球文石成核速率高于其它物相(Li et al., 2002)。然而,在离子条件pH 一致的条件下,方解石由于单分子体积更小,吸附位密度应该更大,界面能应该越低。这与纯水体系下方解石比球文石界面能低的结果相一致。因此,加入醇以后,所有碳酸钙物相的界面能都会升高,由于方解石吸附位密度更大,空间位阻效益应该更明显,界面能应该上升更快,有可能实现界面能的反转。文献中(Manoli et al., 2000a)25℃ 温度下10% 体积分数的乙醇-水体系下球文石界面能为41 mJ/m2,低于相同条件下的方解石界面能48.9 mJ/m2,与纯水体系下相反。然而,在本研究的过饱和度高于文献数值(Manoli et al., 2000a),却并没有观察到乙醇—水体系下的球文石析出。这说明,界面能的高低并不是决定球文石是否形成的决定性因素。
另一个可能的解释是多羟基醇与水溶液形成了复杂的氢键网络,而CO32-离子可以与溶剂形成氢键,羟基数量的增多增加了CO32-离子与溶剂之间的氢键强度和数目,从而限制了CO32-离子自由调节的方位。醇—水体系中,随着醇分子羟基数量的增加,可形成的氢键数目增加,乙醇、乙二醇和丙三醇的粘度依次上升(1.10、17.32、946 cP)。在方解石结构中,所有的CO32-离子都相互平行,CO32-离子若要进入晶格必须要调整吸附方位并进行去溶剂化作用。在乙醇 —水体系中,氢键数目低,CO32-离子的溶剂化作用弱,可以较快的实现方位调整和去溶剂化作用。当加入丙三醇后,强大的氢键网络使CO32-离子活动受限,难以去溶剂化实现与晶格中已有CO32-的对齐,强烈抑制了方解石的形成。球文石结构中的CO32-离子平面并不平行且相对最高对称方向有一定角度偏离,降低了晶体结构的对称性(Christy, 2017),有利于CO32-离子进入晶格,从而形成球文石结构。而乙二醇分子具有2个羟基,氢键网络强于水和乙醇但低于丙三醇,所以形成了方解石和球文石的混合物相。
5 结论
本文研究了25℃ 下0%、5%、10% 体积分数乙醇,乙二醇,丙三醇—水溶液中碳酸钙过饱和情况下的矿物物相以及成核诱导时间。研究发现在纯水以及乙醇—水体系下沉淀的碳酸钙矿物物相为单一的方解石;在丙三醇—水体系下为单一的球文石物相;在乙二醇—水体系下为方解石和丙三醇的混合物相。在此基础上标定了25℃下方解石和球文石在醇—水混合体系下的溶解度积,并通过经典成核理论分析了实验获得的诱导时间与方解石、球文石过饱和度的关系。在此基础上,获得了乙醇—水体系下方解石以及丙三醇—水体系下球文石的界面能随醇种类、比例的定量变化关系。研究发现醇的加入会增加方解石和球文石的界面能,增加程度与体积分数呈线性关系。方解石界面能数值小于球文石,丙三醇对球文石界面能的影响略高于乙醇对方解石的影响。醇的羟基定向吸附造成的吸附位密度降低是提升矿物界面能最可能的机制,介电常数的降低也一定程度提升界面能,表面电荷的变化并不是主要原因。界面能大小并不是决定碳酸钙物相的唯一因素,在实验条件范围内,溶剂氢键数量决定了碳酸根的自由度。丙三醇—水溶液复杂氢键网格限制了CO32-平行的方解石结构形成,而对对称度低CO32-不平行的球文石成为动力学优势物相。而水和乙醇-水体系氢键作用弱,界面能低的方解石物相成为热力学优势物相。在富羟基有机物的生物环境中,球文石的形成具有动力学上的优势,高的界面能有利于团簇的聚合生长,而球文石低有序结构易于低活度CO32-的沉淀。球文石可作为方解石或文石的前驱碳酸钙储库快速沉淀,通过溶解再沉淀形成最终的方解石或文石生物骨骼。
猜你喜欢
宝藏(2021年8期)2021-09-15
农业研究与应用(2021年2期)2021-08-12
烟草科技(2021年6期)2021-06-24
——详解淄博文石皴纹及赏石文化
宝藏(2021年1期)2021-03-10
发明与创新·中学生(2020年9期)2020-10-15
石油钻探技术(2020年4期)2020-10-09
世界有色金属(2020年4期)2020-05-16
中国烟草学报(2019年5期)2019-11-14
宝藏(2019年9期)2019-09-25
宝藏(2018年12期)2019-01-29
