
C1q肿瘤坏死因子相关蛋白9对大鼠心肌梗死后室性心律失常的影响∗
2021-01-04 02:19魏言昭李威杨双李艳君唐艳红
中国心脏起搏与心电生理杂志 2020年6期
魏言昭 李威 杨双 李艳君 唐艳红
C1q肿瘤坏死因子相关蛋白家族(CTRPs)其他成员与脂联素(APN)具有相似的结构,研究证实,CTRPs除APN 之外,至少包括15个成员,分别是CTRP1-15,与APN 存在惊人相似的组织结构和生化特性。CTRPs的转录几乎完全由脂肪细胞表达,其中人和小鼠组织CTRPs的表达最广泛,CTRP9在人类心脏上高表达,在功能上作为活性心肌保护相关因子。已有证据表明,CTRP9主要通过脂联素受体1(AdipoR1)在心肌梗死(MI),或糖尿病合并MI模型上对心肌细胞和成纤维细胞发挥抗凋亡、抗氧化、抗纤维化等作用,通过内皮细胞发挥舒张血管等作用[1-2]。目前研究证实CTRP9抗心肌重构作用明显,其对心肌电生理是否产生影响,以及是否存在CTRP9直接相关的离子通道途径,未有相关研究。笔者通过Ad-CTRP9在MI大鼠中过表达,观察CTRP9对MI大鼠在体心肌电生理的影响并探讨此影响的机制。
1 材料与方法
1.1 实验动物分组及处理 体重接近的64只健康雄性SD 大鼠,12周龄,体重为(180±20)g,购自武汉大学人民医院实验动物中心。随机分为四组:假手术对照(Control)组,心肌梗死(MI)组,Ad-CTRP9+MI(Ad-CTRP9)组,Ad-GFP+MI(Ad-GFP)组,每组16只。Ad-CTRP9组及Ad-GFP 组分别通过颈静脉给药Ad-CTRP9(上海吉凯基因化学技术有限公司)或Ad-GFP(上海吉凯基因化学技术有限公司),5天后予以MI造模,病毒的浓度为5×109pfu。所有动物均在20~25℃的SPF 级环境内恒温饲养。MI造模时给大鼠腹腔注射60 mg/kg的3%戊巴比妥钠进行麻醉,将其仰卧位固定于手术台上。行气管插管术,连接小动物呼吸机,正压通气,潮气量10~15 ml,呼吸频率70次/分。开胸后用扩胸器固定,充分暴露其右心室游离壁的同时避免损伤肺叶。在肺动脉圆锥左缘与左心耳根部交界处下2 mm 处用6/0线结扎冠状动脉左前降支,以即刻心尖表面颜色变白和心电图Ⅱ导联ST 段抬高判定MI造模成功,心肌表面颜色变白与颜色正常交界区域定义为梗死周边区。假手术组除不结扎外,其他处理同MI造模。
1.2 在体心电图(ECG)记录 MI造模14天后假手术组于相对应时间,每组随机选取10只大鼠进行电生理检查,给予3%戊巴比妥钠(60 mg/kg,腹腔注射)麻醉大鼠后,利用Power Lab 数据采集系统(澳大利亚ADInstruments公司)进行Ⅱ导联体连续记录心电图2 h,利用Powerlab数据分析系统,自动分析和测量RR 间期、QT 间期和校正的QT(QTc)间期(采用Bazett算法计算QTc),测定的时域线性指标包括RR 间期,所有窦性RR 间期的标准差(SDNN),频谱分析包括低频(LF)(nu)、高频(HF)(nu)及LF/HF。
1.3 心室电生理检查 MI造模14天后假手术组于相应时间,每组随机选取10只大鼠进行电生理检查,大鼠称重后腹腔注射3%的戊巴比妥钠(30 mg/kg)充分麻醉,经腹腔注射肝素500 u/kg,肝素化10 min后自胸骨剑突下剪断两侧肋骨暴露胸腔,迅速分离剪断主动脉。剪下心脏后先置于4℃改良台式液中去除残余血,然后将主动脉套在Langendorff心脏灌流装置上。以新鲜K-H 液逆行恒温(37℃)、恒压(5.86k Pa)灌流心脏15 min,K-H 液以95% O2+CO2混合气体,灌注流速控制为8 ml/min。2根电极分别连接肺动脉圆锥及心尖部记录心电图,通过BL-420F 生物机能实验系统(澳大利亚ADInstruments公司)观察记录分析心律失常。
单相动作电位(MAP)记录:将刺激电极置于梗死周边区,假手术组于相对应位置给予固定频率起搏(脉宽2 ms方波,电压5V),在LabChart7.0记录分析软件中选择稳定连续的8个MAP 波形,经软件自动计算得出各部位单相动作电位复极至20%、50%和90%时程(APD20,APD50和APD90)。
采用S1S2程序刺激的方法测量心室有效不应期(VERP):S1∶S2=8∶1,即8个S1发放一次S2刺激,S1S1起搏周长为350 ms,刺激电压为8 V。S1S2从200 ms以步长10 ms逐步递减。当出现S2不能夺获心室时,则从上一个S1S2为起始以2 ms步长递减开始测量,将不能夺获心室的最长S1S2间期定义为该部位有效不应期(ERP)。S1S1起搏周长(PCL)从150 ms到30 ms,以10 ms连续递减,直到出现MAP电交替(ALT)。每个PCL 持续30 s,然后休息30 s再进行下一次刺激。ALT 定义为在连续10次MAP的振幅至少相差5%。
室性心律失常(VA)诱发:经5 min恢复自发搏动以待消除心电记忆效应后,保持相同条件,间断给予刺激电压5 V、频率为50 Hz、持续时间2 s、连续单向方波的猝发型串刺激(Brust刺激),每次进行10次重复Brust刺激,刺激之间正常心律恢复时间>1 min。记录VA 发生类型、持续时间,计算室性心律失常诱发率(Burst刺激诱发出室性心动过速、心室颤动的动物数除以总数)。
1.4 心肌组织CTRP9、KChIP2 和Kir2.1 蛋白检测 MI造模14天后假手术组于相应时间,利用蛋白质免疫印迹法测定心肌组织中CTRP9、KChIP2和Kir2.1的表达量。每组随机选取6只大鼠,用剪刀剪取梗死周边区心肌组织,置于组织匀浆器中,加入总蛋白提取液,匀浆5~20 min直到组织充分破碎,然后冰上放置10~20 min 后,再匀浆5~20 min,将匀浆液吸出放到1.5 ml离心管中。以9 000 rpm,4℃离心10 min,取适量上清置于新的1.5 ml离心管中,提取细胞总蛋白,使用BCA 蛋白质浓度测定试剂盒(北京艾德来)测定样品蛋白浓度。将蛋白质样品与对应的SDS上样缓冲液混合,100℃,加热10 min。将凝胶板放入电泳槽中,吸取样品并加样。设置电压为80 V 进行凝胶电泳,30 min后观察电泳情况。调整电压为100 V,继续电泳2 h,再次观察电泳情况,直到溴酚蓝达至胶板下沿约1 cm处,结束电泳;SDS-PAGE 电泳分离蛋白后将蛋白转入PVDF膜上。移入相应离子通道抗体稀释液中,在4℃下孵育过夜。然后在膜的蛋白面侧加入新配制的ECL 混合溶(A∶B=1∶1),在暗室中曝光、显影、定影。AlphaEaseFC 软件分析目标带的光密度值。
1.5 统计学分析 所有数据运用SPSS 23.0软件进行分析。正态分布的计量资料以¯x±s 表示,同组自身前后比较采用配对t 检验,同组自身多个时点的比较用重复测量资料的方差分析。以P<0.05为差异有显著性。
2 结果
2.1 四组心肌组织CTRP9的表达水平 与Control组相比,MI组心肌中CTRP9表达水平显著降低;与Ad-GFP组相比,Ad-CTRP9组CTRP9表达水平升高并显著高于Control组,P 均<0.05),表明腺病毒成功转染CTRP9并且转染效率高。如图1和表1所示。
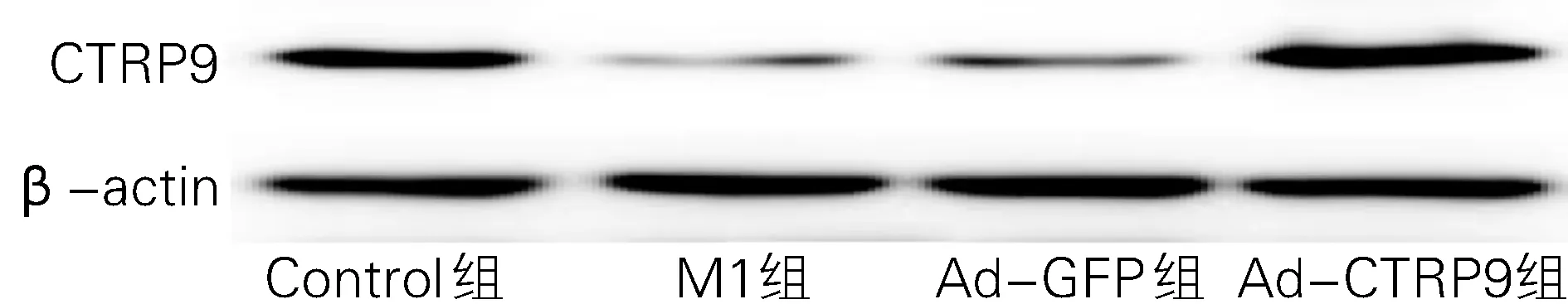
图1 CTRP9在心肌组织中的表达量

表1 心肌组织中CTRP9的表达量及KChIP2和Kir2.1在心肌组织中的表达量
2.2 ECG 参数和心室HRV 的变化 与Control组相比,MI组和Ad-GFP组大鼠QT间期、QTc间期和QRS时限均延长,而与Ad-GFP组比较,Ad-CTRP9组大鼠QT间期、QTc 间期和QRS波时限显著缩短(P 均<0.05);与Control组相比,MI组SDNN 均下降,而与Ad-GFP 组比较,Ad-CTRP9 组SDNN显著提高;与Control组相比,MI组LF 及LF/HF值显著增加,HF(nu)值显著降低;而Ad-CTRP9组这一趋势减弱,LF 及LF/HF 值降低,HF 值增加,并使之趋于正常。如表2和表3所示。
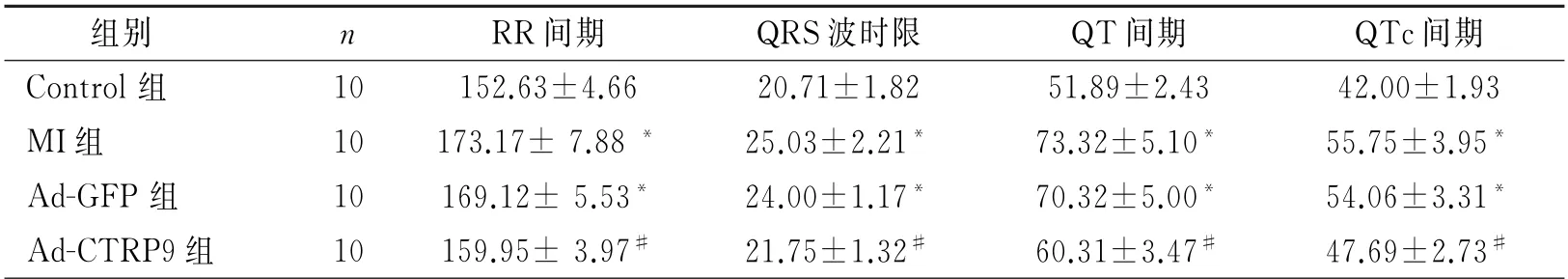
表2 四组心电指标的比较/ms
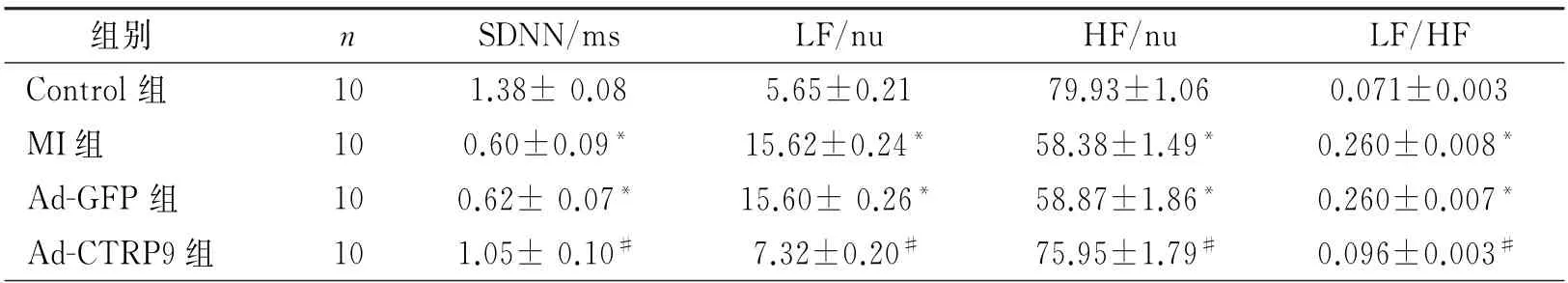
表3 四组心率变异性的比较
2.3 四组心室ERP和APD 的比较 与Control组相比,MI 后 梗 死 周 边 区ERP、APD20、APD50和APD90显 著 延 长,相 反,与Ad-GFP 组 比,Ad-CTRP9组ERP、APD20、APD50和APD90显著缩短(P 均<0.05)。如表4所示。
2.4 ALT周长的比较 四组均诱发出APD电交替,梗死周边区APD电交替周长如图所示,与Control组相比,MI组梗死周边区APD 电交替周长显著延长,与Ad-GFP组比较,Ad-CTRP9组诱发阈值提高,梗死周边区电交替周长缩短。如表4和图2所示。

表4 四组心电指标的比较/ms
2.5 VA 诱发率的比较 与Control组相比,MI组的诱发率显著增加(10/10 vs 0/10,P <0.05),而Ad-CTRP9组较Ad-GFP组的诱发率下降(3/10 vs 10/10,P<0.05)。
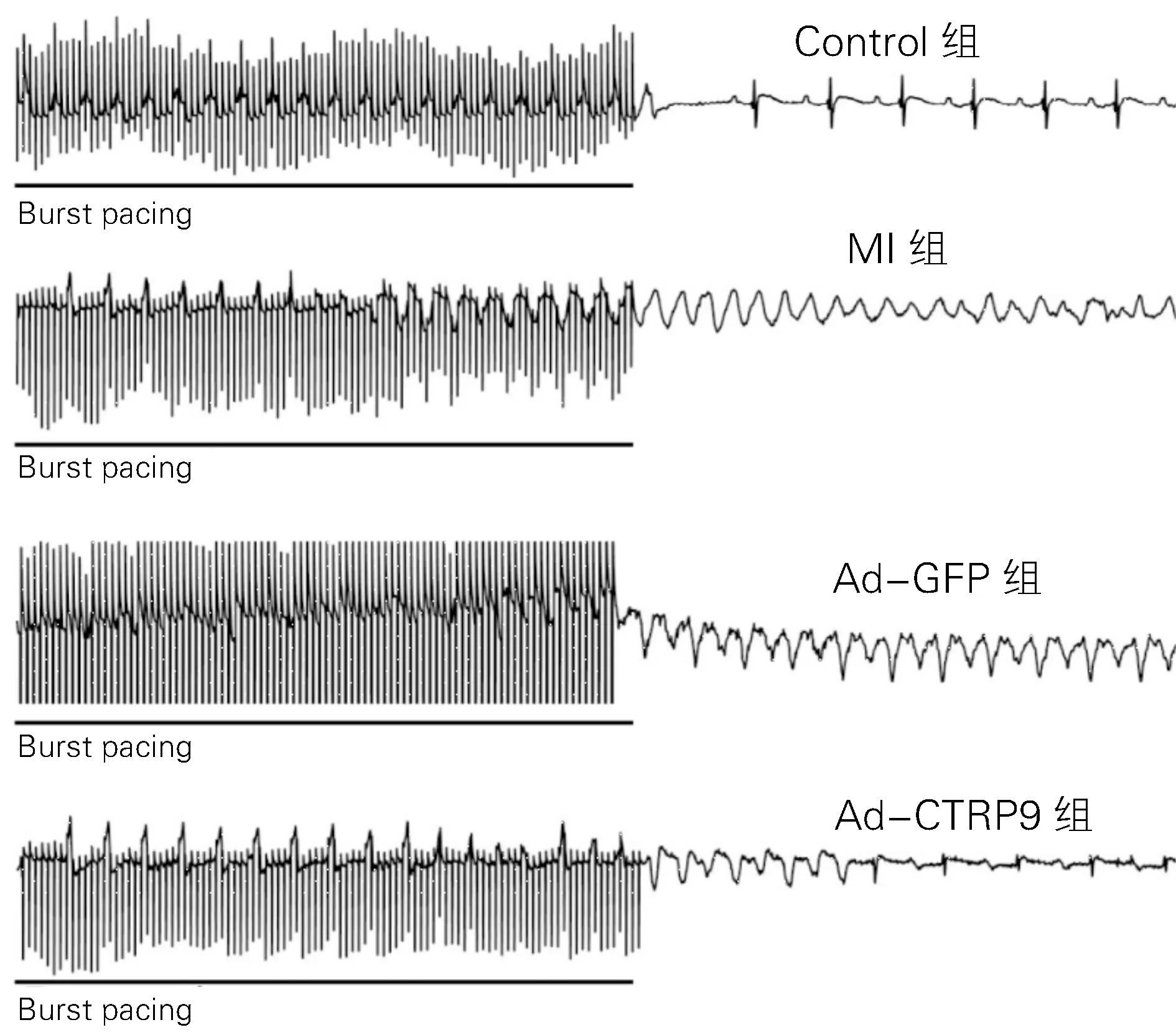
图3 四组Burst刺激诱发VA 典型图
2.6 四组KChIP2和Kir2.1蛋白的比较 与Control组相比,MI组KChIP2和Kir2.1蛋白的表达显著降低,Ad-CTRP9组KChIP2和Kir2.1蛋白高于Ad-GFP组。如图4和表1所示。
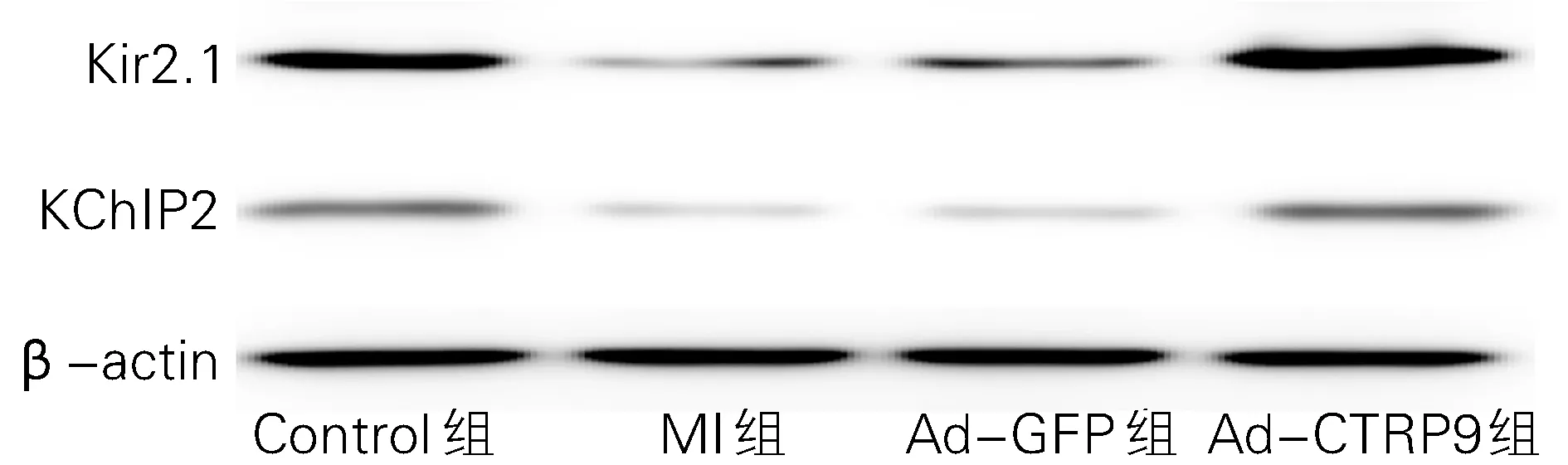
图4 各组心肌组织中KChIP2的表达量
3 讨论
心脏电重构是对MI后结构和功能改变的应激反应,电重构的特征之一是复极化异常,特别是APD 的延长[3],既往研究表明,复极化的电异质性是诱发VA 的主要原因。MI后电生理特性的透壁梯度(包括兴奋性,APD 和心肌不应性的分散)为折返提供了基础,折返是心律不齐的主要机制。在本研究中,笔者发现MI后,梗死周边区的心室ERP和APD 延长,心室易损性增加,CTRP9可明显改善MI后ERP、APD 延长和心室易感性,提示CTRP9可改善MI后的电重构。心脏重构的离子机制涉及外向K+电流(Ik),内向Ca2+电流(ICa)和内向晚Na+电流(INa)之间的复杂相互作,除了改变电流密度外,Ik、ICa和INa的空间分布也发生了变化,这些变化改变了心脏的正常复极化梯度,可能导致心律失常的发生[4]。既往研究表明,各种K+电流,如快速外向瞬时钾电流(Ito),快速激活的延迟整流钾电流(Ikr)和延迟整流钾通道(Iks)在梗死周边区心肌细胞中表达下调[5],促进VA 的发生。
KChIP2基因缺陷导致Ito完全丧失,易发生室性心动过速,KChIP2是一种KChIPs家族的Ito通道相互作用蛋白,作为Kv通道的辅助亚基,对Kv通道的正常功能发挥了重要作用[6]。Kir2.1是IK1的成孔α亚基,IK1是静息电位和动作电位后期复极化的主要决定因素,研究表明IK1的下调导致心力衰竭后的致命性心律失常[7],MI后心律失常的发生与静息电位的降低和IK1表达的减少紧密相关[5-8],适当增强IK1而使静息电位超极化有利于消除异常的自律性。IK1的抑制降低了复极化时有效不应期与APD 的比率,因此降低了复极化后的心肌不应性,表明抑制IK1可能导致HF 或阿斯综合征后出现的心律失常,促进IK1的表达可能降低这种折返性心律失常的易感性。本研究中CTRP9 过表达能缓解MI大鼠梗死周边区KChIP2 和Kir2.1 的表达减少,因而上调Ito,IK1并减少VA 的发生。本研究证实CTRP9 能增加梗死周边区心肌中KChIP2 和Kir2.1的表达,或许是一种有效的抗心律失常策略。
猜你喜欢
蔬菜(2022年9期)2022-09-23
中国典型病例大全(2022年10期)2022-05-10
中国心脏起搏与心电生理杂志(2022年1期)2022-03-03
现代中西医结合杂志(2021年34期)2021-12-22
家禽科学(2021年10期)2021-11-22
实用心电学杂志(2021年5期)2021-10-26
心电与循环(2021年5期)2021-10-11
保健与生活(2021年11期)2021-06-10
昆明医科大学学报(2021年2期)2021-03-29
世界中医药(2019年11期)2019-09-10
