
基于加权基因共表达网络的膀胱尿路上皮癌预后及疗效相关基因筛选
2020-12-31 06:58:22李佳丽曾梓航刘嘉彬孟详喻
转化医学杂志 2020年6期
李佳丽,曾梓航,刘嘉彬,张 冉,李 硕,孟详喻,李 胜,李 伟
膀胱尿路上皮癌(bladder urothelial carcinoma,BLCA)占膀胱癌发病率的90%以上,是泌尿系统中最常见的恶性肿瘤之一[1-2]。BLCA具有多发性、复发率高等特点,其发病机制尚未完全明确。目前有研究表明,BLCA的发病机制可能和多基因、多层次的细胞信号网络紊乱有关[3]。BLCA的治疗效果(即进展、稳定、部分缓解、完全缓解)显著地影响了患者的预后,且其基因层面的机制有待阐明。
加权基因共表达网络分析(weighed gene co-expression network analysis,WGCNA)是一种构建基因共表达网络的系统生物学算法,该算法以转录组数据为分析基础,在世界生物医学范围均得到广泛的应用[4]。WGCNA作为生物网络分析中最具代表性的分析方法[5],可以多层次多数量地分析生物系统中基因表达的信息,已经为多物种的基因表达分析提供了具有临床意义的结果。由于目前缺少关于BLCA的治疗效果及预后的基因层面机制的系统大数据研究。因此,本研究开展了WGCNA方法筛选BLCA预后及治疗效果的相关基因。
1 材料和方法
1.1 材料
1.1.1 训练数据集 本研究从癌症基因组图谱(the cancer genome atlas,TCGA)数据库[6]中获取同时具有mRNA测序及相应临床数据的BLCA患者408例作为训练数据集。mRNA测序数据经过RSEM归一化和log2转换[7]。选择年龄、性别、身高、体重、人种、生存时间、吸烟年数、治疗效果、淋巴结浸润、病理分级及TNM分期为临床表型,以生存时间和治疗效果为主要研究结果。
1.1.2 验证数据集 本研究获取基因表达汇编(gene expression omnibus,GEO)数据库中的GSE13507和GSE3167两个BLCA数据集进行验证[8,9]。GSE3167包含9例正常膀胱组织和41例BLCA样本。GSE13057包含68例正常膀胱或癌旁组织和188例BLCA样本,并且含有随访信息。
1.2 方法
1.2.1 WGCNA 本研究采用R语言WGCNA程序包[10]进行WGCNA。一般按照以下步骤进行:①离群值的去除:将患者按照基因表达模式进行层次聚类,并将离群样本剔除;②基因模块的形成:将基因进行K-means聚类,形成模块;③模块筛选:将模块进行主成分分析,即各模块第一主成分与临床表型进行皮尔森相关分析,得到模块与表型的相关系数即模块显著性(module significance,MS)及P值;④模块内重要基因筛选:模块隶属度(module membership,MM)>0.5为阈值,以基因对治疗效果的重要性(gene significance,GS)大小排序,筛选模块中与治疗效果显著相关的基因。
1.2.2 生存分析 本研究利用R语言Survival程序包进行生存分析。将筛选出的基因按表达量大小绘制生存曲线并进行Cox回归分析[11],获得与预后显著相关的基因。
1.2.3 通路活性分析 本研究利用GSCALite(http://bioinfo.life.hust.edu.cn/web/GSCALite/)数据库[12]进行肿瘤相关通路活性的分析。通路包括细胞凋亡、细胞周期、DNA损伤修复、上皮细胞-间充质转化、雄激素受体通路、雌激素受体通路、PI3K/Akt,、Ras/MAPK、RTK及TSC/mTOR。该信息由Rehan Akbani等[13]通过多组学系统生物学分析得到。
1.3 统计学方法 利用R语言3.4.4统计软件。在WGCNA中,相关分析均采用皮尔森检验;肿瘤和正常组织的基因表达量差异使用t检验;生存分析采用单因素Cox回归进行。P<0.05为差异有统计学意义。
2 结果
2.1 WGCNA结果
2.1.1 去除低表达基因和离群样本 将在70%及以上样本中无表达的基因去除后,408个样本进行层次聚类,删去离群的5个样本,剩下403个样本。样本聚类及相应临床表型数据,图1A。
2.1.2 基因模块聚类及模块筛选 在403个样本中进行基因模块聚类,共18 196个基因聚类为89个基因模块(图1B)。paleturquoise模块与生存时间(MS=-0.13,P=0.009)和治疗效果显著相关(MS=-0.27,P<0.000 1),但与年龄无关(MS=-0.058,P>0.05)。因此,本研究选择paleturquoise模块做后续分析。
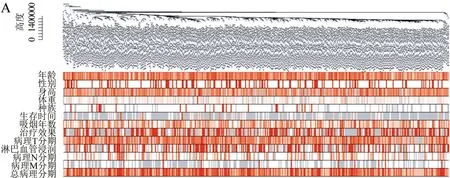
图1 WGCNA
2.1.3 模块内重要基因筛选 在paleturquoise模块中,选取GS值排序前20的基因(表1),进行单因素Cox回归分析。结果显示:8个基因GPHN、CPSF2、PRMT5、EIF2S1、KLC1、SLC39A9、SRP54及CNIH为预后相关因子。
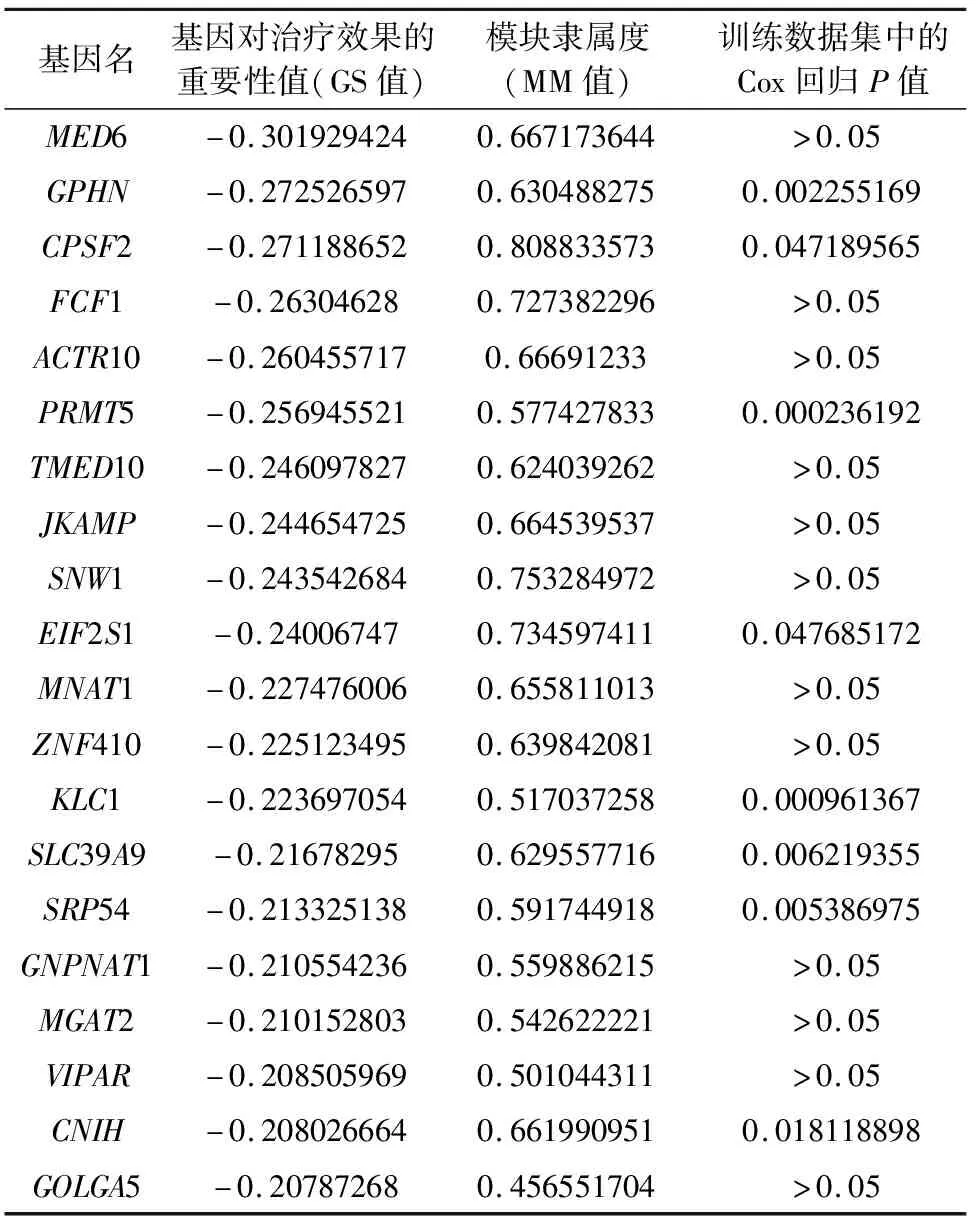
表1 GS值前20基因的信息
2.2 多数据集验证
2.2.1 关键基因在肿瘤和正常组织中的表达 本研究用GEO数据库中的GSE3167和GSE13507分析关键基因在肿瘤和正常组织中的表达(图2A)。大多数基因在肿瘤和正常组织中显示出了差异表达。
2.2.2 关键基因与预后的关系 将上述8个关键基因按照表达量高低分为两组,并绘制生存曲线。与预期一致,这8个基因在TCGA数据中均为预后的不利因素(图2B)。在GSE13507中趋势与TCGA一致,除CPSF2和SLC39A9之外,其他基因均为预后的风险因素(图2C)。
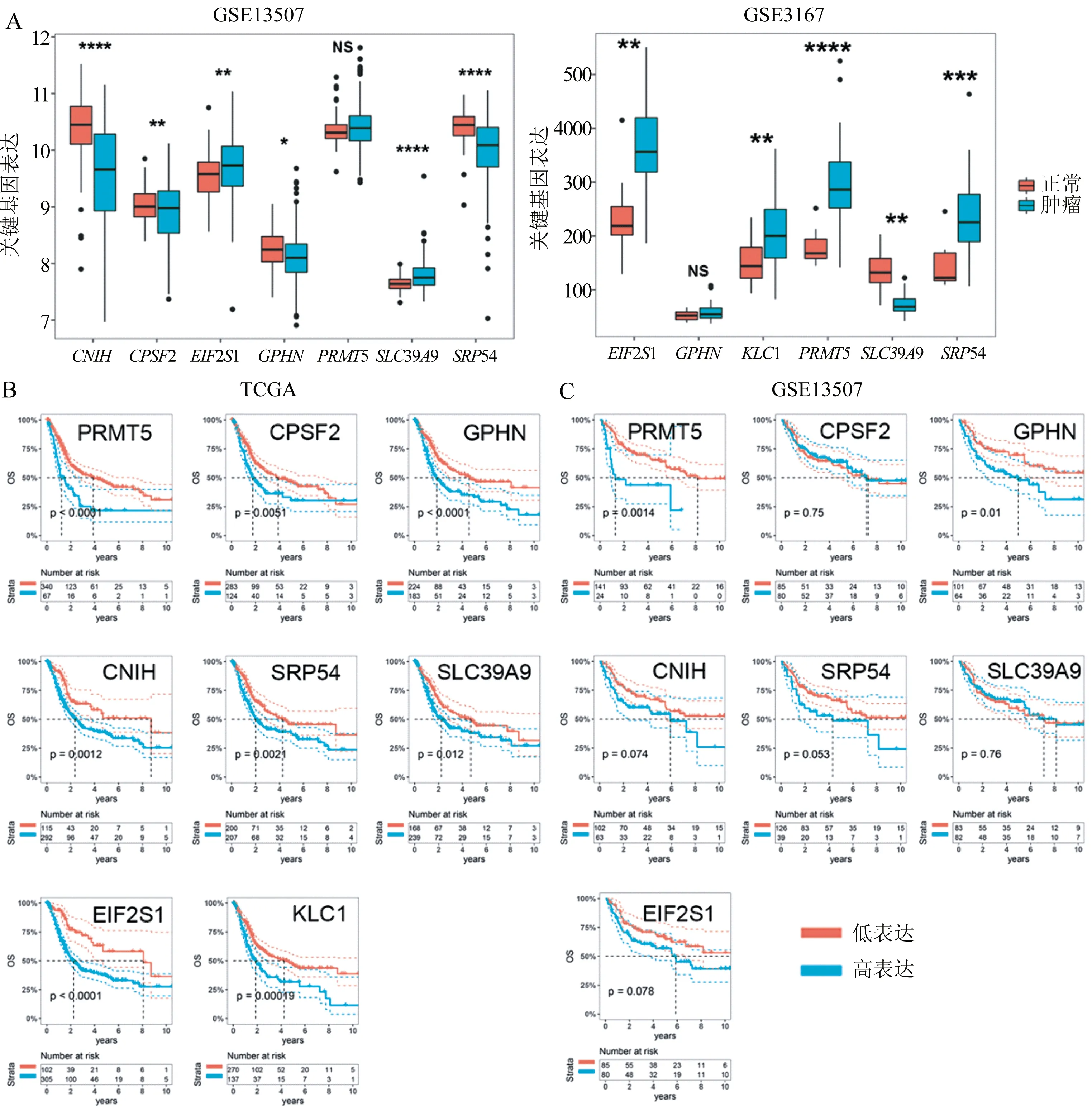
图2 关键基因的多数据集验证
2.3 关键基因与肿瘤通路活性有关 通路活性分析显示GPHN、CPSF2、PRMT5、EIF2S1、KLC1、SLC39A9、SRP54及CNIH在10个肿瘤相关通路中均显示出一定活性(图3)。几乎所有的基因都对受体酪氨酸激酶(receptor tyrosine kinase,RTK)有一定活性。活性较强的基因通路关系还有CPSF2、EIF2S1、PRMT5对细胞周期,EIF2S1对细胞凋亡,KLC1对EMT和TSC/mTOR通路。
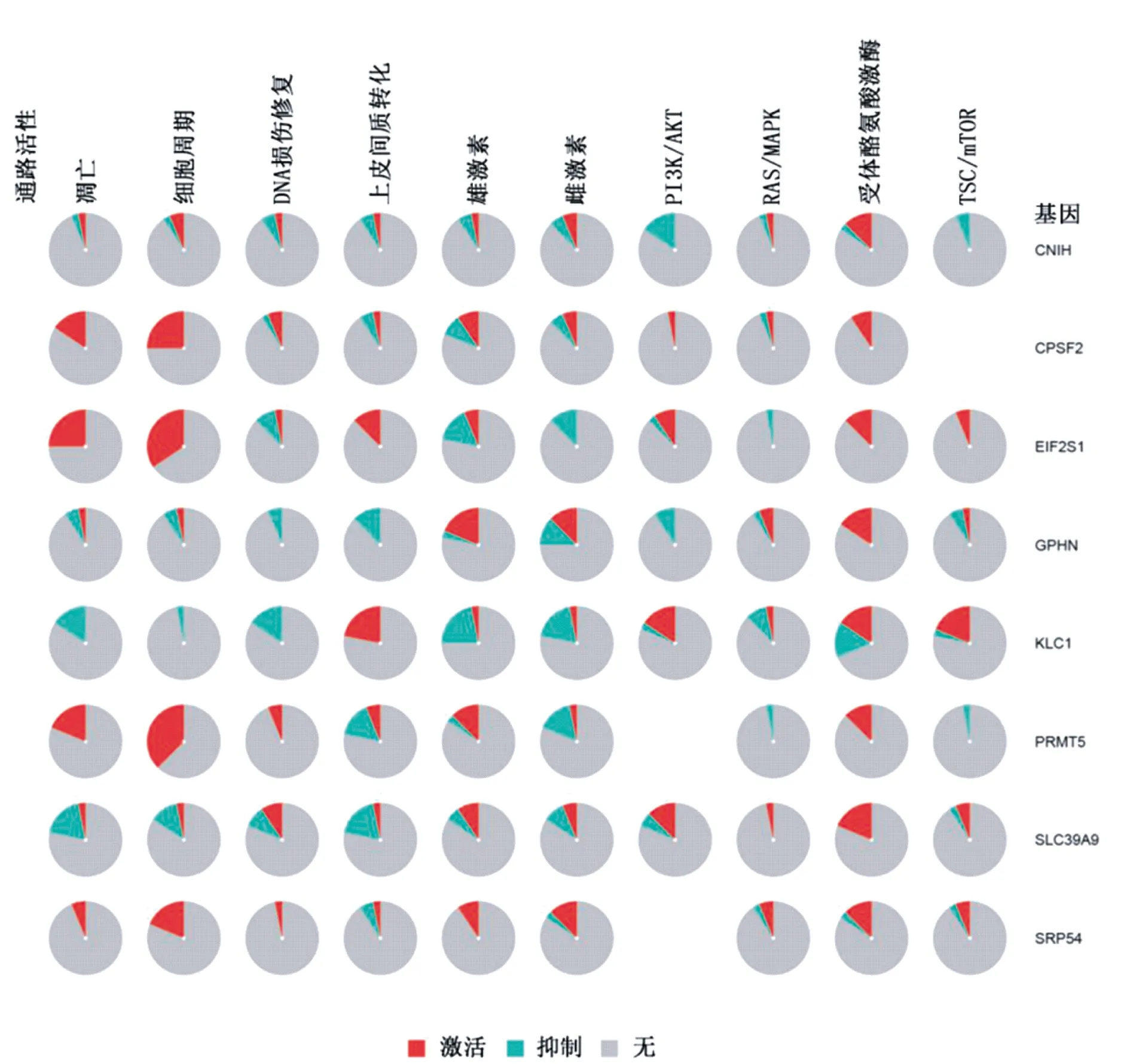
图3 通路活性分析
3 讨论
本研究对408个临床患者样本信息进行WGCNA,构建加权基因共表达网络,从89个基因模块中筛选与患者疗效和生存均显著的paleturquoise模块,并且该模块与年龄无关。通过设定MM值的阈值,以GS值大小排序,与生存显著相关的基因取交集,最终筛选出与预后及治疗效果显著相关的GPHN、CPSF2、PRMT5、EIF2S1、KLC1、SLC39A9、SRP54和CNIH基因。通过对上述基因的肿瘤相关通路活性分析,发现上述基因都有一定激活RTK通路的作用。GPHN是参与膜蛋白细胞骨架的微管相关蛋白[14];CPSF2主要与mRNA剪接和RNA聚合酶Ⅱ转录终止有关[15];PRMT5编码属于甲基转移酶家族的酶,编码的蛋白质催化甲基基团转移到氨基酸精氨酸中,包括组蛋白、转录延伸因子和肿瘤抑制因子p53[16];EIF2S1是翻译起始因子EIF2的亚基,与蛋白质合成有关[17];KLC1编码驱动蛋白轻链家族的成员,与微管运动活性有关[18];SLC39A9与金属离子转运及Nrf2途径有关[19];SRP54与mRNA剪接有关,并且具有GTP酶活性[20];CNIH是AMPA受体辅助蛋白,涉及TGF-α家族蛋白的选择性转运和成熟[21]。
目前有不少文献将WGCNA方法运用到多种肿瘤的研究中,如肾肿瘤[22]、前列腺癌[23]、肺腺癌[24]、乳腺癌[25]及肠癌[26]等。也有研究关注膀胱癌的WGCNA,但与本研究的方法及结果均有所不同。李丹妮[27]的研究关注女性特异性膀胱癌相关基因,Deng等[28]的研究关注肿瘤特异性基因,Li等[29]的研究关注肿瘤TNM分期。本研究关注预后及治疗效果,更注重于临床意义。因为本研究对于预后相关基因的筛选使用了WGCNA与Cox回归相结合的方法,对于模块的筛选及其他影响因素如年龄进行了限制,所以所得结果更具有稳健性。
本研究同样具有局限性:①只筛选了paleturquoise模块,可能会漏掉同样有显著意义的其他模块里的基因;②多数据集中,这些基因在BLCA和正常组织中的表达并不完全一致,这可能与样本的异质性以及样本组织中肿瘤细胞的纯度有关;③关于上述基因在BLCA作用的研究还相当少,样本量有限,机制仍不明确。因此,未来还需要更多的研究来加以验证。
本研究发现GPHN、CPSF2、PRMT5、EIF2S1、KLC1、SLC39A9、SRP54及CNIH基因与BLCA的疗效和预后均显著相关,并且8个基因都对RTK有一定关联;其中,CPSF2、EIF2S1、PRMT5对细胞周期有较大相关性,EIF2S1对细胞凋亡,KLC1对EMT和TSC/mTOR通路都有较大的关联性。它们有望成为BLCA新的预后标记物或潜在治疗靶点,为未来的基础和临床研究提供了新的思路。
猜你喜欢
中学生数理化·高一版(2021年2期)2021-03-19 08:32:00
知识经济·中国直销(2018年8期)2018-08-23 09:16:16
电子测试(2017年15期)2017-12-18 07:19:27
数学学习与研究(2017年3期)2017-03-09 18:12:42
中国老区建设(2016年1期)2016-02-28 09:32:00
智能系统学报(2015年4期)2015-12-27 09:38:39
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
医学研究杂志(2015年3期)2015-06-10 06:41:52
电子设计工程(2015年6期)2015-02-27 12:04:53
创业家(2015年1期)2015-02-27 07:52:02
