
极紫外光刻胶产气的定性和定量检测
2020-12-25 02:53陈金平郝青山王双青杨树敏赵俊吴衍青曾毅于天君杨国强李嫕
分析化学 2020年12期
关键词:产气
陈金平 郝青山 王双青 杨树敏 赵俊 吴衍青 曾毅 于天君 杨国强 李嫕

摘 要 利用压强升高法建立了极紫外(Extreme ultraviolet, EUV)光刻胶产气检测系统, 对以分子玻璃(Molecular glass)螺芴(9,9′-Spirobifluorene, SP)为主体材料的光刻胶薄膜体系Film A、B、C和D进行产气的定性和定量分析, 其中, Film A的主体材料外围取代基团为叔丁氧羰基(t-Butylcarbonyl, Boc), Film B和C是在Film A光刻胶薄膜顶层覆盖不同厚度的保护层, Film D的主体材料外围取代基团为醋酸金刚烷酯(Adamantyl acetate, Ad)。采用四极杆质谱检测光刻胶薄膜在EUV曝光条件的产气组分, 结果表明, Film A产气的主要来源为光照产生的酸催化光刻胶主体材料脱Boc取代基反应释放的异丁烯(C4H8)和CO2气体, 以及少量由于产酸剂(Photo-acid generator, PAG)分解释放的苯类挥发性组分。覆盖保护层的Film B、C产气成分与Film A类似, 但各离子峰的丰度明显降低。Film D的质谱图上显示气体释放成分为CO2和极微量的金刚烷类取代基的碎片峰。通过高精度真空规定量分析不同薄膜的产气量, 原位实时检测结果表明, 光刻胶薄膜产气速率在曝光初始阶段最快, 而后逐渐变缓或趋于稳定, 表明光刻胶薄膜表面的分子在EUV光照更容易释放气体。对比Film A和Film B、C体系发现, 顶层覆盖可以显著降低光刻胶的产气速率和产气量, 增加顶层覆盖厚度, 抑制产气效果更明显, 在Film A顶层覆盖厚度30 nm的保护层, 10 mJ/cm2 曝光剂量下, 产气量从1.19×1015 molecule/cm2降低到2.35×1014 molecule/cm2, 降低了约5倍, 证明顶层覆盖是降低光刻胶薄膜产气的有效方法。不同取代基团的主体材料形成的光刻胶薄膜Film D和Film A在EUV曝光中的产气差别明显, Film D的产气速率和产气量比Film A降低了10倍以上, 表明光刻胶主体材料的外围取代基团对光刻胶薄膜的产气量具有显著影响, 更大分子量和更高脱保护反应活化能的取代基团有助于降低光刻胶薄膜的产气量。
关键词 光刻胶; 产气; 分子玻璃; 极紫外; 光刻
1 引 言
近四十余年, 随着光刻技术的不断进步, 半导体器件的工艺节点已经从最早的10 μm缩小到7 nm, 目前, 国际上重要的半导体公司(如三星公司、台积电公司)已经在7 nm节点工艺中导入极紫外(Extreme ultraviolet, EUV, 13.5 nm)光刻技术, 即将量产的5 nm节点中关键工艺也采用EUV光刻技术, EUV光刻已成为最新一代光刻技术。光刻胶和光刻技术的发展相互匹配, 开发能满足EUV光刻技术要求的光刻胶材料是目前面临的一个重要挑战[1]。EUV光刻技术与其它光刻技术不同, 使用波长13.5 nm的光源, 要求在超高真空的条件下进行曝光, 由于EUV光子能量高, 光刻胶的主体材料和各种辅助材料在曝光过程中会分解释放小分子化合物, 这些小分子化合物在超高真空中呈气态, 会对EUV曝光机或曝光系统中的精密光学元件造成污染和损坏[2~4]。由于EUV曝光机制造成本高昂, 曝光过程中需要尽可能降低光刻胶产气导致的曝光机污染, 产气量成为设计和研发EUV光刻胶材料时需要考虑的重要因素, 防止光刻胶材料产气或降低产气量, 保护EUV曝光机或曝光系统, 成为评价光刻胶的重要指标, 也是评价EUV光刻胶实用性的标准。
目前, 对于EUV光刻胶产气量的评估方法仍处于实验室阶段, 尚没有成熟的商品化产气检测设备。Chauhan[5]和Dentinger[6]等最早采用石英晶体微天平(Quartz crystal microbalance, QCM)结合质谱分析方法研究各种聚合物光刻胶在EUV光照下的产气, 该方法对设备精度和灵敏度要求高, 尤其是对于微量的产气, QCM的检测限并不能完全达到实验要求。Watanabe等[7,8]通过质谱定性分析光刻胶产气种类和来源, 但无法进行定量檢测。 Santillan等[9~11]提出利用EUV曝光前后曝光腔内气体压强的变化量分析产气总量的方法, 同时结合质谱分析产生气体的种类、性质和来源等。利用上述这些方法对不同光刻胶的研究结果表明, 光刻胶组分、取代基团、产酸剂含量等对EUV曝光后的产气量都有显著影响[12~17], 基于这些研究结果, 发展了一些抑制或平衡光刻胶产气的方法[18,19]。本研究组联合上海同步辐射光源, 在EUV光刻线站设计构建了一套EUV光刻胶产气的检测系统, 并利用此系统对分子玻璃(Molecular glass)光刻胶产气量进行了检测, 结果表明, 分子玻璃骨架对于EUV光照具有良好的稳定性, 产气主要来源于取代基团的酸催化分解反应, 通过调整配方, 提高防酸扩散剂比例, 降低取代基团数目, 可以有效降低产气量[20]。最近, 本研究组对多个系列的分子玻璃光刻胶的光刻性能进行了研究和评价, 获得了多种高分辨率、低边缘粗糙度、高灵敏度的综合性能优良的光刻胶材料和体系[21,22], 但对材料的产气性能和影响因素还有待进一步研究和评价。本研究对4种光刻胶薄膜产气情况进行了定性和定量检测, 研究主体材料结构以及光刻胶薄膜顶层覆盖对产气的影响, 为EUV光刻胶材料的设计和曝光工艺优化提供指导。
2 实验部分
2.1 仪器与试剂
ProsmaPlus TM四极杆质谱仪(德国普发公司); PBR-260高精度真空规(德国普发公司); SP-30溅射离子泵(日本三井公司); CEE200X匀胶机、CEE1300X热台(美国Brewer Science公司); SE200-BM光谱型椭偏仪(美国AST公司); H-1硅片预处理系统(河北神通光电科技有限公司)。 水溶性聚(4-磺酸盐)-苯乙烯高分子聚合物(分析纯, Sigma-Aldrich); 六甲基二硅烷胺(Hexamethyl disilylamine, HMDS, 电子级, 北京科华微电子材料有限公司)。
2.2 实验方法
光刻胶涂膜: 先将硅片置于HMDS预处理系统中进行疏水处理, 处理后的硅片放在匀胶机上, 根据所需要的薄膜的厚度选择不同的转速进行旋涂, 光刻胶主体材料的浓度范围控制在20~40 mg/mL, 光刻胶涂膜转速通常选择2000 r/min, 得到均匀薄膜, 热台前烘坚膜, 前烘温度为100℃, 时间为180 s, 坚膜后的薄膜厚度在光谱型椭偏仪上进行测试; 光刻胶层坚膜后, 再进行覆盖层的旋涂, 旋涂转速为3000 r/min。曝光剂量根据光通量、光子能量以及曝光时间进行计算。
3 结果与讨论
3.1 分析方法和体系的建立
EUV曝光条件下的光刻胶产气检测系统建在上海同步辐射光源软X射线干涉光刻线站上, 整个检测系统主要包括进样腔、曝光腔、四极杆质谱仪(Quadropole mass spectrometer, QMS)以及高精度真空规(如图1)。通过QMS质谱仪对曝光过程中产生的气体种类进行分析, 实现对曝光过程中气体的原位、实时、定性检测, 通过高精度真空规实时检测腔体真空度, 并根据光照前后气压的变化对产生气体的总量进行计算, 实现对产气量的定量检测, 定性结合定量的方法可以实现对不同光刻胶体系产气的全面评价。
为研究主体材料中取代基团的结构对光刻胶产气的影响, 选用的光刻胶主体材料结构为本研究组前期已经进行光刻工艺评价的螺芴(9,9′-Spirobifluorene, SP)结构。以SP为骨架, 外围取代基团分别为叔丁氧羰基(t-Butylcarbonyl, Boc)和醋酸金刚烷酯(Adamantyl acetate, Ad)两种基团[22]。选择不同厚度的亲水性聚合物覆盖光刻胶薄膜, 研究顶层覆盖对产气的影响。光刻胶薄膜类型如表1所示, 其中Film A、B和C的光刻膠配方完全相同, 均为含有Boc取代基团的主体材料复配相同的产酸剂和防酸扩散剂, 不同之处在于Film B和C覆盖厚度分别为15和30 nm的顶层保护(Topcoating)膜。Film D和A均为不含有顶层保护层的光刻胶薄膜, 但光刻胶主体材料不同, 分别为Boc和Ad取代基团的SP主体材料, 两者的产酸剂和防扩散剂的配方比例完全相同。
3.2 气体种类的定性分析
首先对光刻胶薄膜样品在无光照条件下进行空白实验, 即将光刻胶样品放到曝光腔体后, 在EUV光照之前, 检测系统内达到平衡时的微量气体组分。在高真空度条件下, 空白背景在无光照时的质谱如图2A所示。曝光腔体内主要气体组分为残留的极微量空气, 质谱实时检测到的主要成分为H2O (m/z 18)、N2 (m/z 28), 以及少量化碎片离子峰O (m/z 16)、OH (m/z 17), 与不加入样品时的质谱图基本相同, 说明在无光照条件下, 光刻胶薄膜释放的挥发性气体量很少, 质谱检测限范围内基本检测不到新的气体产生。
通过EUV照射光刻胶样品, 质谱实时检测释放出的气体, 扣除空白背景后得到不同类型的光刻胶薄膜释放气体结果如图2B~2E所示, 分别对应Film A、B、C和D的产气情况。与未曝光时的空白质谱图对比可见, Film A、B、C和D产生气体的分子离子峰的数量和丰度都发生了变化。Film A中产生的气体组分的分子离子峰在m/z 28处的丰度显著增加, 并增加了新的分子离子峰, 分别为m/z 39、40、41、44、55和56, 以及微量的m/z 77和78。结合主体材料和产酸剂的结构以及文献[23,24]报道, 推测质量范围位于28~56的分子离子峰应为产酸剂在光照下产生酸, 此酸催化主体材料发生了脱取代基Boc反应而形成的分子离子峰和碎片峰。分解机理如图3A所示, 主要分子离子峰分别为异丁烯C4H8(m/z 56)和CO2(m/z 44), 其它峰m/z 28、39、41和55则为异丁烯的碎片峰。同时, 在EUV光照下, 产酸剂产酸过程中会发生三苯基硫鎓盐的分解反应, 释放出少量苯类气体, 在质谱图上观察到微量的苯分子离子峰C6H6(m/z 78)和对应碎片峰C6H5(m/z 77), 这与产酸剂的产酸分解过程完全吻合。进一步对照实验证明, 当直接光照不含产酸剂的主体材料薄膜时, 并没有明显的产气现象, 表明EUV光照直接分解主体材料产气并不明显, 因此, Film A产气的主要来源是酸催化主体材料脱取代基反应以及产酸剂自身的分解反应。
Film B和C是在Film A的薄膜上分别覆盖15和30 nm的防产气高分子涂层, 光照产气对应的质谱图与Film A相比, 增加的主要分子离子峰仍为主体材料酸分解而产生的CO2 (m/z 44)和异丁烯C4H8(m/z 56), 但丰度随高分子覆盖层厚度增加而显著降低; 在Film B中可以观察到的丰度极低的苯分子离子峰(m/z 77, 78), 但在Film C已经几乎观察不到苯分子离子峰, 异丁烯C4H8(m/z 56)的丰度也变得很弱, 可见高分子覆盖层对产气起到了明显的抑制作用。覆盖材料本身对EUV光子的吸收导致光刻胶曝光量降低, 也是产气量降低的可能原因之一。Film D的外围取代基团由Boc变为Ad, 其酸催化反应机理与Boc不同(图3B), 质谱上显示的分子离子峰也完全不同。通过与Film A质谱图对比可知, 产生气体的分子离子峰的丰度显著降低, 且组分发生了改变, 新产生的主要组分为CO2 (m/z 44), 可能来自反应后羧酸的分解, 同时观察到了少量的金刚烷取代基的碎片峰(m/z 50~56; m/z 91~93)以及极微量产酸剂分解的苯分子离子峰(m/z 77~79)。综上, 以Ad为取代基团的主体材料形成的光刻胶薄膜(Film D)经EUV曝光后产气量比相应的Boc取代基团的主体材料形成的Film A产气量少。
3.3 产气的定量计算和分析
采用Santillan等[9]已报道的压强升高法定量检测EUV曝光后光刻胶产气量。在高真空度下, 光照光刻胶薄膜产生的气体会导致曝光腔内的真空度降低, 通过测量曝光前后的腔内压强差可以定量检测光刻胶薄膜的曝光产气量。EUV曝光腔内为高真空 (约10-6 ~10-7 Pa) 条件, 产生的气体可以近似为理想气体状态, 根据理想气体状态方程, 可以采用公式(1)和(2)计算最大产气速率和产气量:
公式(1)中, NR为EUV光刻胶产生气体的速率(molecules/(cm2·s)), ΔPMAX是腔内压强的最大升高值(Pa), Se是真空离子泵的抽气速率(m3/s), Na是阿伏加德罗常数(6.022×1023 molecules/mol); 公式(2)中, ND为产气总量(molecules/cm2), ΔPi是在起始检测时间ts (s)到终止检测时间tD (s)之间腔内特定时刻i时的真空度变化(Pa), Δt是EUV光照时间(s), A是光刻胶样品的曝光面积(cm2)。两个公式中, R为气体常数(8.314 Pa·m3/(K·mol)), T为真空腔内温度 (K)。通过公式(1)和(2), 结合压强的升高变化, 即可检测出光刻胶的产气速率NR和某个时间段(或曝光剂量范围)的产气总量ND。
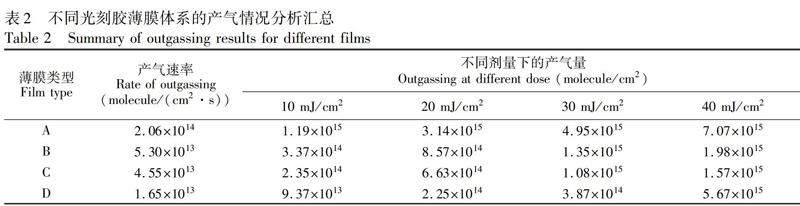
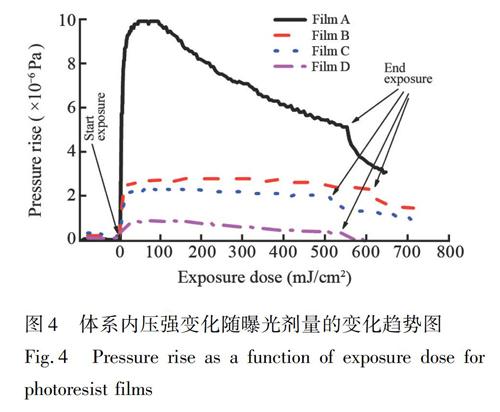
以系统稳定后的压强为初始压强, 开始光照初始剂量(或时间)为零点, 记录体系压强随剂量(或光照时间)的变化, 可以准确获得体系压强升高值与曝光剂量之间的关系曲线。图4为4种光刻胶薄膜在EUV曝光过程中, 曝光腔内压强随曝光剂量的变化曲线。光照开始阶段产气速率明显较快, 曝光剂量在达到约40 mJ/cm2后, 放气速率开始变慢或趋稳; 曝光剂量达到500 mJ/cm2后, 停止光照, 产气速率出现显著降低。曲线的变化趋势表明, 产气主要来自曝光初始阶段, 可能是因为光刻胶薄膜表面的分子光照更容易发生反应并释放气体, 在光刻胶薄膜上层增加高分子覆盖层(Film B 和C), 体系的气压升高值显著低于未覆盖的光刻胶薄膜(Film A), 而且顶层覆盖厚度越大, 气压升高越小, 这也进一步表明表面的分子更容易反应释放气体。根据最大产气速率公式(1), 可以计算出各光刻胶薄膜体系的最大产气速率, 以Film A为例, 其中相对于初始系统压强最大增加值△Pmax为9.87×10-6 Pa, 离子泵抽气速率Se为160 L/s (真空度10-5~10-6 Pa条件下), 根据曝光光斑测得曝光面积A为1.87 cm2, 温度T为室温298 K, 通过计算可以得到Film A的最大产气速率NR为2.06×1014 molecule/(cm2·s)。利用相同的方法可以得到Film B、C和D的最大产气速率NR分别为5.30×1013 molecule/(cm2·s)、455×1013 molecule/(cm2·s)和1.65×1013 molecule/(cm2·s), 產气速率大小顺序为: Film A>Film B>Film C>Film D。根据公式(2)可以计算出某曝光剂量(或者某时刻)下的产气量。根据图4中所对应的曝光剂量与产气速率的积分可以计算出当曝光剂量分别为10、20、30和40 mJ/cm2时, 不同组分光刻胶薄膜体系的产气量见表2, Film A、B、C和D的产气量均随曝光剂量增加而增大, 在相同曝光剂量下, 四种光刻胶薄膜产气量的变化趋势与产气速率大小变化趋势一致。
通过比较Film A、B和C产气量结果, 在10 mJ/cm2曝光剂量下, 产气量从1.19×1015 molecule/cm2降低到2.35×1014 molecule/cm2, 降低约5倍, 因此, 光刻胶表面进行顶层覆盖, 是有效抑制光刻胶薄膜产气的一种方法。对比Film A和D的产气量可知, 主体材料的取代基团对于光刻胶薄膜的产气量具有显著影响, Film A和D具有同样的分子骨架, 只是外围取代基团不同, 但Film D的产气量比Film A的产气量降低了10倍以上, 一方面可能与主体材料发生酸催化脱保护基反应后, 生成的组分分子量更大, 不容易挥发有关; 另一方面可能与Film D主体材料取代基团酸催化反应活化能更高, 需要更高的反应温度才能实现脱保护基团的反应有关[25]。
4 结 论
利用建立的光刻胶产气检测系统, 对以螺芴(SP)结构为主体材料的光刻胶薄膜体系Film A、B、C和D进行产气的定性和定量分析。 质谱分析结果表明, 在EUV曝光条件下, Film A、B和C光刻胶薄膜产气的主要来源是酸催化的主体材料脱Boc取代基反应释放的异丁烯以及CO2, 还有少量产酸剂自身的分解反应释放的气体, Film D由于取代基团不同, 主要来源为CO2和极微量的金刚烷取代基的碎片峰。定量分析结果表明, 光刻胶薄膜产气在曝光初始阶段速率最快, 顶层覆盖可以显著降低光刻胶的产气速率和产气量, 增加顶层覆盖厚度, 抑制产气的效果更明显, 证实顶层覆盖是降低光刻胶产气的有效方法。外围修饰醋酸金刚烷酯取代基团的光刻胶薄膜Film D在EUV曝光过程中, 产气速率和产气量比Film A降低了10倍以上, 表明光刻胶主体材料的外围取代基团对于光刻胶薄膜的产气量具有显著影响, 更大分子量和更高脱保护反应活化能的取代基团有助于降低光刻胶薄膜的产气量。本研究结果对于EUV光刻胶主体材料的设计和探索抑制光刻胶产气的方法具有重要的指导作用。
References
1 Li L, Liu X, Pal S, Wang S L, Ober C K, Giannelis E P. Chem. Soc. Rev., 2017, 46(16): 4855-4866
2 Chen J Q, Louis E, Lee C J, Wormeester H, Kunze R, Schmidt H, Schneider D, Moors R, van Schaik W, Lubomska M. Bijkerk F. Opt. Express, 2009, 17(19): 16969-16979
3 Lee S, Doh J G, Lee J U, Lee I, Jeong C Y, Lee D G, Rah S Y, Ahn J. Curr. Appl. Phys., 2011, 11(4): S107-S110
4 Koida K, Niibe M. Appl. Surf. Sci., 2009, 256(4): 1171-1175
5 Chauhan M M, Nealey P F. J. Vac. Sci. Technol. B, 2000, 18(6): 3402-3407
6 Dentinger P M. J. Vac. Sci. Technol. B, 2000, 18(6): 3364-3370
7 Watanabe T, Hamamoto K, Kinoshita H, Hada H, Komano H. Jpn. J. Appl. Phys., 2004, 43(6B): 3713-3717
8 Watanabe T, Kinoshita H, Sakaya N, Shoki T, Lee S Y. Jpn. J. Appl. Phys., 2005, 44(7B): 5556-5559
9 Santillan J J, Kobayashi S, Itani T. Jpn. J. Appl. Phys., 2008, 47(6): 4922-4925
10 Santillan J J, Kobayashi S, Itani T. Proc. SPIE, 2008, 6923: 692342
11 Santillan J J, Toriumi M, Itani T. Proc. SPIE, 2007, 6519: 651944
12 Shiobara E, Takagi I, Kikuchi Y, Sasami T, Minegishi S, Fujimori T, Watanabe T, Harada T, Kinoshita H, Inoue S. J. Photopolym. Sci. Technol., 2015, 28(1): 103-110
13 Sugie N, Takahashi T, Katayama K, Takagi I, Kikuchi Y, Tanaka H, Shiobara E, Inoue S. Proc. SPIE, 2013, 8679: 86792E
14 Ho G H, Shao C H, Sung J J, Kang F H, Kao C B, Hung W L, Chou Y L, Huang Y H. J. Vac. Sci. Technol. B, 2012, 30(5): 051602
15 Pollentier I, Lokasani R, Gronheid R. J. Photopolym. Sci. Technol., 2012, 25(5): 609-616
16 Sugie N, Takahashi T, Katayama K, Takagi I, Kikuchi Y, Shiobara E, Tanaka H, Inoue S, Watanabe T, Harada T, Kinoshita H. J. Photopolym. Sci. Technol., 2012, 25(5): 617-624
17 Takahashi T, Sugie N, Katayama K, Takagi I, Kikuchi Y, Shiobara E, Tanaka H, Inoue S, Watanabe T, Harada T, Kinoshita H. Proc. SPIE, 2012, 8322: 83221E
18 Sakamoto R, Fujitani N, Onishi R, Nishita T. J. Photopolym. Sci. Technol., 2013, 26(5): 685-689
19 Chang S H, Chen S F, Chen Y Y, Chien M C, Chien S C, Lee T L, Chen J J H, Yen A. Proc. SPIE, 2013, 8679: 86790O
20 Chen L, Xu J, Yuan H, Yang S M, Wang L S, Wu Y Q, Zhao J, Chen M, Liu H G, Li S Y, Tai R Z, Wang S Q, Yang G Q. Sci. China Chem., 2014, 57(12): 1746-1750
21 Peng X M, Wang Y F, Xu J, Yuan H, Wang L Q, Zhang T, Guo X D, Wang S Q, Li Y, Yang G Q. Macromol. Mater. Eng., 2018, 303(6): 1700654
22 Chen J P, Hao Q S, Wang S Q, Li S Y, Yu T J, Zeng Y, Zhao J, Yang S M, Wu Y Q, Xue C F, Yang G Q, Li Y. ACS Appl. Polym. Mater., 2019, 1(3): 526-534
23 Kudo H, Suyama Y, Oizumi H, Itani T, Nishikubo T. J. Mater. Chem., 2010, 20(21): 4445-4450
24 Kudo H, Niina N, Sato T, Oizumi H, Itani T, Miura T, Watanabe T, Kinoshita H. J. Photopolym. Sci. Technol., 2012, 25(5): 587-592
25 Tarutani S, Tsubaki H, Fujimori T, Takizawa H, Goto T. J. Photopolym. Sci. Technol., 2014, 27(5): 645-654
Qualitative and Quantitative Measurement of Outgassing
of Molecular Glass Photoresists under Extreme
Ultraviolet Lithography
CHEN Jin-Ping1, HAO Qing-Shan1, WANG Shuang-Qing2, YANG Shu-Min3, ZHAO Jun3,
WU Yan-Qing3, ZENG Yi1,4, YU Tian-Jun1, YANG Guo-Qiang*2,4, LI Yi*1,4
1(Key Laboratory of Photochemical Conversion and Optoelectronic Materials,
Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China)
2(Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Photochemistry,
Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China)
3(Shanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute,
Chinese Academy of Sciences, Shanghai 201204, China)
4(University of Chinese Academy of Sciences, Beijing 100039, China)
Abstract A system was set up for quantitative and qualitative analysis of outgassing of photoresists using pressure rise method. The outgassing of molecular glass photoresist films (Film A, B, C and D) based on 9, 9′-spirobifluorene (SP) derivatives was evaluated by the system. Film A was formed by SP with substituent group of tert-butyloxycarbonyl (Boc), and Film B and C were prepared by covering Film A with top-coating material in different thicknesses. Film D was formed by SP with substituent group of adamantyl acetate (Ad). The components of residual gas during the EUV irradiation were detected by the quadruple mass spectrometer (QMS). In the case of Film A, it showed that the source of the outgassing was mainly from the acid catalytic reaction of substituent of Boc group, releasing isobutylene (C4H8) and CO2 gas, along with a small amount of benzene component produced by the decomposition of photo acid generator (PAG). Film B and C, covered with protective layer, were similar to that of film A, but the ion abundance decreased obviously. Ion peak of CO2 molecule and the fragment peaks of adamantane substituent were observed in mass spectra of Film D. The amount of outgassing for different films was analyzed in-situ by an ion gauge, and it was found that the rate of outgassing was fast in the initial stage of exposure, and then slow down, suggesting that the molecules on the surface of the films were more likely to release gas. Besides, the top-coating could significantly reduce the rate of outgassing comparing the results of Film A and Film B/C, for instance, the outgassing could be reduced from 1.19×1015 molecule/cm2 to 2.35×1014 molecule/cm2 under the exposure dose of 10 mJ/cm2 by covering with 30-nm thickness of top-coating material, suggesting that the top-coating was an effective method to reduce outgassing. By comparing the outgassing of Film D and Film A that was formed by the resist materials with different peripheral substituent groups, it was found that the outgassing of Film D was decreased by 10 times than that of Film A, indicating that the substituent group of the photoresist materials had a significant effect on the outgassing, and the substituent group with higher molecular weight and thermal activation energy (Ea) was useful to reduce outgassing.
Keywords Photoresist; Outgassing; Molecular glass; Extreme ultraviolet; Lithography
(Received 24 June 2020; accepted 27 October 2020)
This work was supported by the National Science and Technology Major Project of the Ministry of Science and Technology of the People′s Republic of China (Nos. 2018ZX02102005, 2011ZX02701).
2020-06-24收稿; 2020-10-27接受
本文系國家科技重大专项(Nos. 2018ZX02102005, 2011ZX02701)资助
* E-mail: gqyang@iccas.ac.cn; yili@mail.ipc.ac.cn
猜你喜欢
时代汽车(2022年3期)2022-02-18
文萃报·周二版(2020年45期)2020-12-23
汽车实用技术(2020年16期)2020-09-06
现代农业科技(2018年22期)2018-01-15
创新时代(2017年8期)2017-08-30
发明与创新·大科技(2017年8期)2017-08-17
农民科技培训(2014年4期)2014-04-23
农业工程技术·农业信息化(2009年1期)2009-02-27
