
基于深层原油物性模拟的分子力场优选及验证
2020-12-24 09:39:36张英男李汝传于顺昌郭牧之燕友果郭文跃
中国石油大学学报(自然科学版) 2020年6期
张英男, 李汝传, 于顺昌, 郭牧之, 燕友果, 郭文跃, 张 军
(1.中国石油大学(华东)材料科学与工程学院,山东青岛 266580; 2.中国石油大学(华东)石油工程学院,山东青岛 266580)
深层油气[1-5]成为油气勘探与增产增储的重要领域[6-8]。盆地深层高温高压条件普遍,形成了由气态烃、石油蒸汽、液态油等构成的复杂流体系统,其流体物性多变、相态特征复杂[9]。分子动力学模拟方法可细致刻画研究对象的物化性质[10-16],如密度、黏度和界面张力等。当前研究中考察的温度、压力条件太低(185~373 K[10]; 343~373 K, 18~30 MPa[11]; 300 K, 0.1 MPa[12]; 303~323 K, 0.1 MPa[13]; 283~333 K, 10 MPa[14]; 293~373 K, 14.5~21.16 MPa[15]; 295.15 K, 0.1 MPa[16]),远低于深层的真实温度和压力条件,并且研究中采用各种不同的分子力场(OPLS[11,14,15]; PCFF[12]; COMPASS[13]; CVFF[16])。笔者采用分子动力学(MD)模拟与从头算分子动力学(AIMD)模拟相结合的计算方法,考察深层原油物性特征和分子力场成键性质,优选适用于深层高温高压条件下的分子力场,验证分子力场的可靠性。
1 原油组分表征和分子力场
1.1 原油组分的实验表征
选取塔里木盆地顺北地区为研究区块,研究区位于顺托果勒低隆起北部,原油密度分布在0.792~0.831 g/cm3,平均值为0.803 g/cm3,呈现出低密度、低黏度的特征,属于轻质油。所用原油样品取自顺北1-2H井,取样井深7 469~7 569.47 m,检测仪器为Varian 3800气相色谱仪。原油样品的轻烃色谱分析(图1)结果表明,直链烷烃质量分数为35.27%,支链烷烃质量分数为24.96%,环烷烃质量分数为19.04%,芳香烃质量分数为9.63%,原油组分的定性定量结果见表1。
在检测所得的组分中,正庚烷、正辛烷、正壬烷和正癸烷的质量分数均超过6.4%,支链烷烃中2-甲基庚烷质量分数为2.91%,环烷烃中甲基环己烷的质量分数为4.05%,芳香烃中间二甲苯的质量分数为2.88%,常温常压下是气体物质的正丁烷的质量分数为0.7%,这些物质可以作为原油样品中的典型组分。本文中选取正辛烷、2-甲基庚烷、甲基环己烷、间二甲苯和正丁烷5种典型组分作为研究对象,考察它们的物性特征。
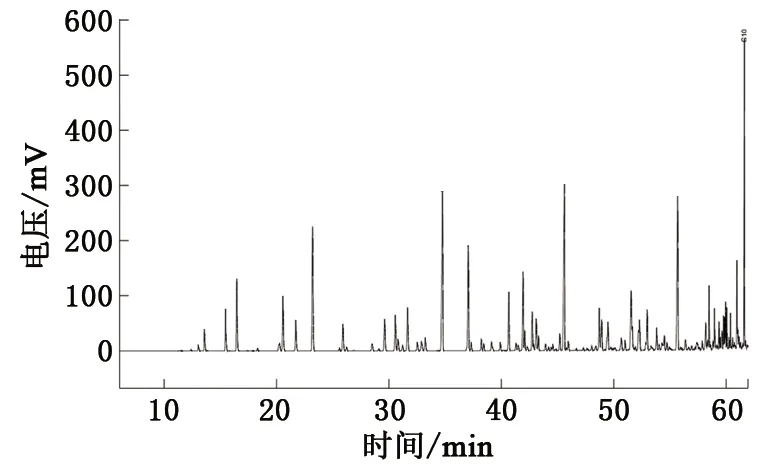
图1 顺北1-2H井原油样品轻烃色谱检测结果Fig.1 Light hydrocarbon chromatography test results of crude oil sample from well Shunbei 1-2H
1.2 分子力场
分子动力学模拟是利用经典牛顿第二定律求解体系内各个粒子随时间的运动轨迹,其中粒子所受的力由体系的势能决定,其关键是得到不同体系的势能函数,而描述体系中原子间相互作用力的势能函数,即为分子力场[17]。分子力场可以看作各类型势能的总和,其数学表达式为
U=Ubond+Uangle+Utorsion+Uvdw+Uel+Uχ.
(1)
式中,Ubond为键伸缩势能;Uangle为键角弯曲势能;Utorsion为二面角扭转势能;Uvdw为范德华非键结势能;Uel为库伦静电势能;Uχ为离平面振动势能。
对于有机分子体系,常见的分子力场有OPLS力场[18]、CVFF力场[19]、COMPASS力场[20]、PCFF力场[21]。本文中选取上述4种典型分子力场,对其准确性、可靠性进行考察,优选出适用于深层高温高压模拟的分子力场。
2 分子动力学模拟
2.1 模型构建
首先,利用Materials Studio软件分别构建正辛烷、2-甲基庚烷、甲基环己烷、间二甲苯和正丁烷的分子结构。然后,利用软件中的Amorphous Cell Construction模块分别构建5种典型组分的模拟盒子(图2),每种组分的模拟盒子尺寸均为5 nm×5 nm×5 nm。对每种组分的模型分别设置OPLS、CVFF、COMPASS和PCFF力场所对应的力场参数及电荷参数,即完成5种组分模拟体系共计20个模型的构建。本文的可视化结果采用VMD软件[22]实现,并生成所需的界面构型图。
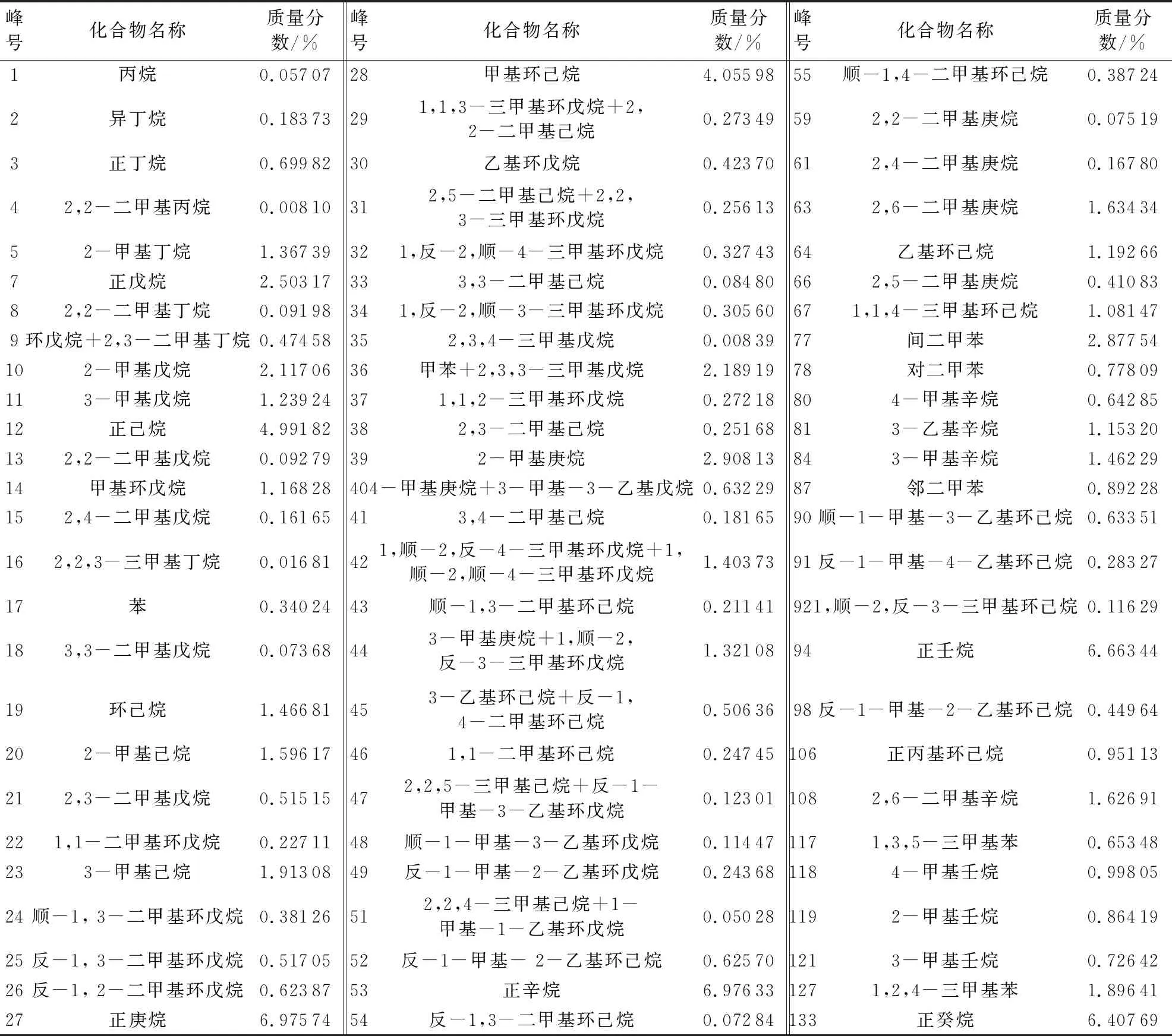
表1 顺北1-2H井原油样品轻烃组分及其质量分数Table 1 Compound and its mass fraction of crude oil sample from well Shunbei 1-2H
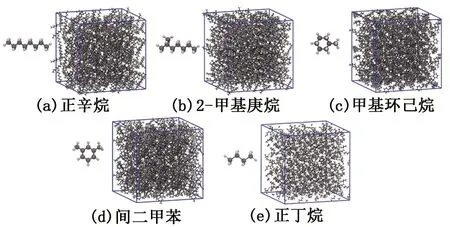
图2 样品的模拟盒子Fig.2 Simulated boxes of samples
2.2 模拟设置
所有的分子动力学模拟均使用LAMMPS软件[23]执行。首先采用最速下降法优化初始构型,对体系能量最小化。然后设置模拟时间步长为1 fs,采用 NPT系综进行3 ns的分子动力学模拟,模拟的前2 ns用于平衡体系,后1 ns用于统计分析数据。模拟时的温度和压力根据研究区实际条件进行设置,钻井、测井和录井资料表明,研究区及附近区块的地层温度分布在373~473 K,主体温度在433 K,地层压力分布在70~120 MPa,主体压力在80 MPa(图3)。模拟盒子采用三维周期性边界条件,截断半径为1.2 nm,长程静电力使用PPPM方法计算。
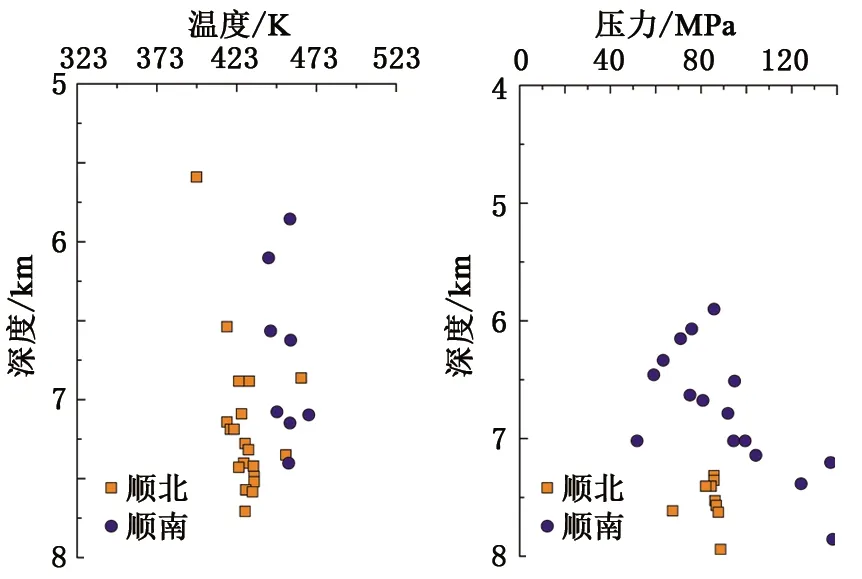
图3 研究区温度和压力特征Fig.3 Characteristics of formation temperature and pressure in research area
3 分子力场优选
3.1 密度验证
考察5种原油组分体系的密度特征,采用AP1700物质物性平台[24]中提供的物性数据与模拟结果进行对比,用于验证各个分子力场的精度。AP1700包含超过2 000种物质的物性参数,其选取超过100万个数据与美国国家标准与技术研究院发布的数据[25]进行对比测试,其数据几乎完全吻合。笔者对比AP1700平台与NIST所发布的正辛烷和正丁烷的密度及黏度数据,发现二者在不同温度、压力条件的数据完全一致,但因NIST未发布2-甲基庚烷、甲基环己烷和间二甲苯的物性数据,故本文中选择AP1700物质物性平台所提供的数据作为5种原油组分物性验证的标准。模拟中采用LAMMPS软件的空间分层方法(ave/chunk)计算密度,模拟盒子中每间隔0.5 Å为一层,每2 ps输出一次计算结果,统计模拟最后1 ns的密度平均值。基于研究区的温度和压力条件,对原油组分的密度特征进行两种模式的考察,分别在不同温度和恒定压力(80 MPa)以及不同压力和恒定温度(433 K)的条件下开展模拟。
恒定压力和变化温度条件下,分子动力学模拟后的密度结果如图4所示。由图4看出,不同分子力场下的密度结果均与AP1700物性数据呈现相同的变化趋势,即密度随温度的升高而不断降低。对于正辛烷、2-甲基庚烷、甲基环己烷和间二甲苯4种物质,OPLS和COMPASS力场的计算结果与AP1700数据十分接近,而CVFF和PCFF力场的计算结果存在一定的偏差。对于正丁烷,4种分子力场的计算结果与AP1700数据均存在偏差,其中CVFF和PCFF力场相对较好,OPLS 和COMPASS力场略差。恒压变温条件下,基于模拟结果统计了5种组分在4种分子力场下的平均误差,OPLS力场的平均误差为2.23%,COMPASS力场为2.28%,CVFF力场为2.85%,PCFF力场为3.20%。
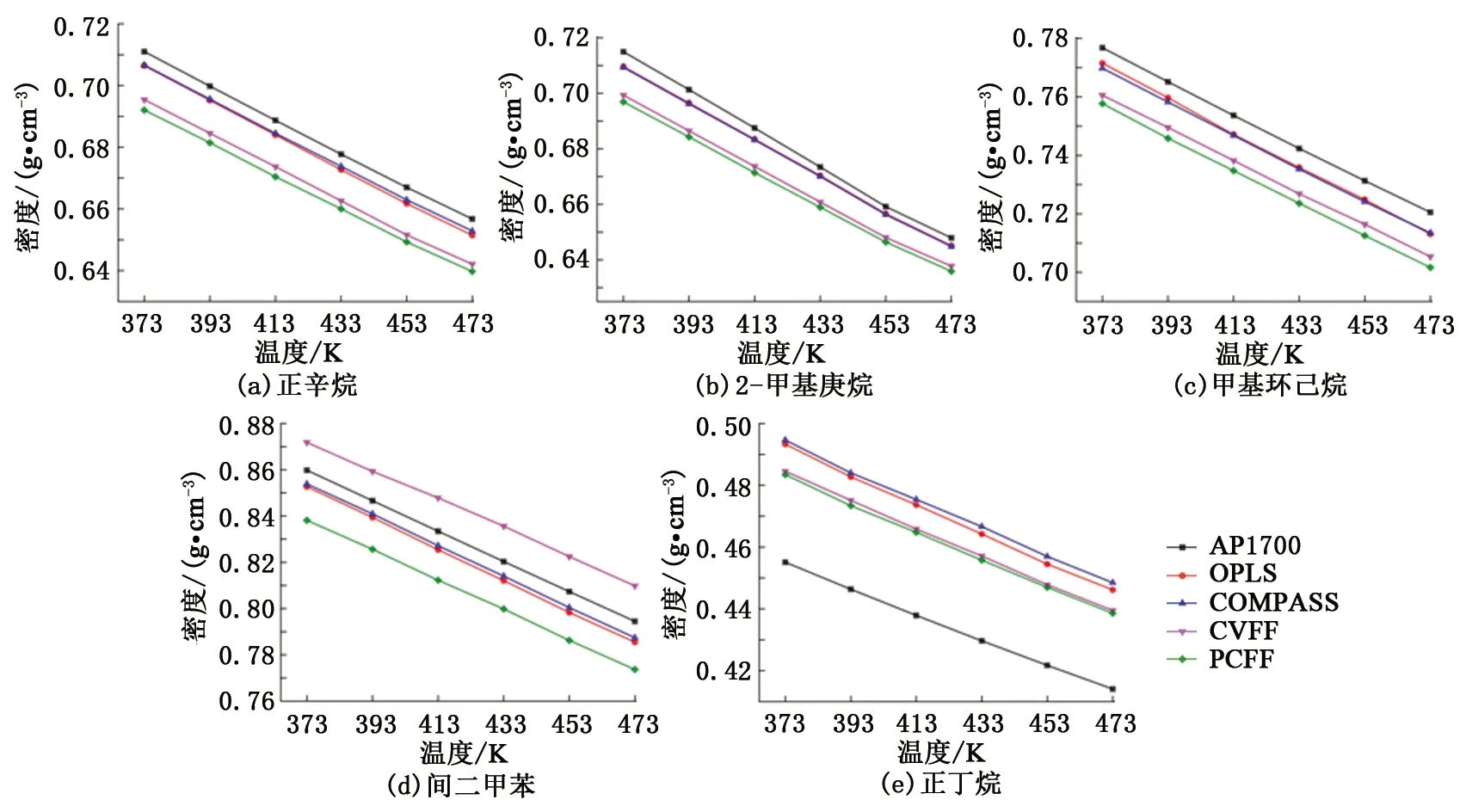
图4 恒定压力和变化温度下样品的密度特征Fig.4 Density characteristics of samples under constant pressure and varying temperature
恒定温度和变化压力条件下的密度模拟结果如图5所示。可以看出,不同分子力场下的密度结果也均与AP1700物性数据呈现相同的变化趋势,即密度随压力的升高而不断增大。变压条件与变温条件下的密度模拟结果相似,OPLS和COMPASS力场对于正辛烷、2-甲基庚烷、甲基环己烷和间二甲苯4种物质的密度模拟更为准确,而CVFF和PCFF力场对于正丁烷的密度模拟相对更好。恒温变压条件下,根据模拟结果统计了5种组分在4种分子力场下的平均误差,OPLS力场的平均误差为2.04%,COMPASS力场为2.08%,CVFF力场为2.71%,PCFF力场为2.98%。不同温度和压力条件下的密度计算结果表明,OPLS力场与AP1700物性数据间的平均误差最小,该力场能够较为准确地反映模拟对象的密度特征。但从误差值来看,其他3种分子力场与OPLS力场间的差异很小。
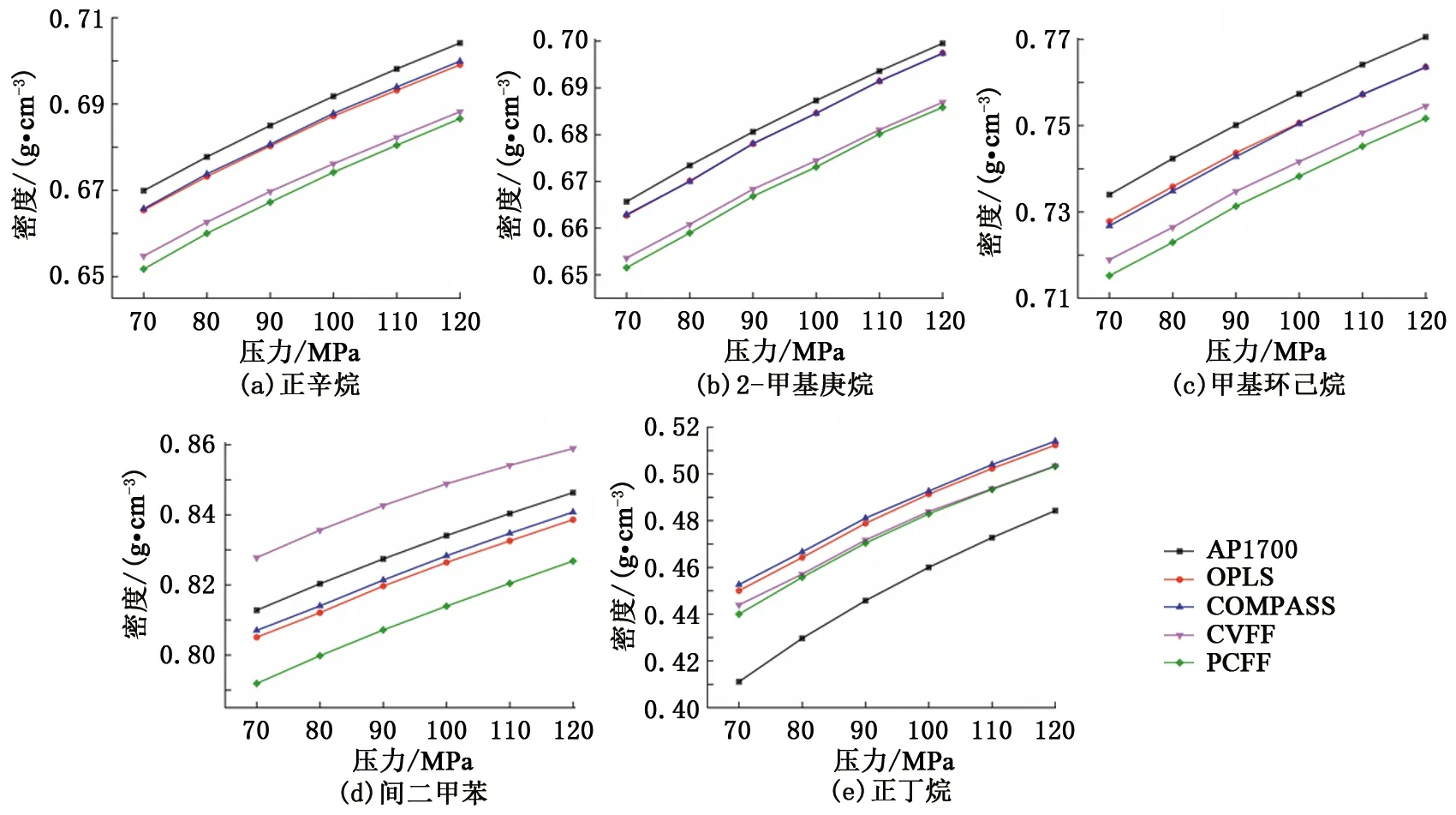
图5 恒定温度和变化压力下样品的密度特征Fig.5 Density characteristics of samples under constant temperature and varying pressure
3.2 黏度验证
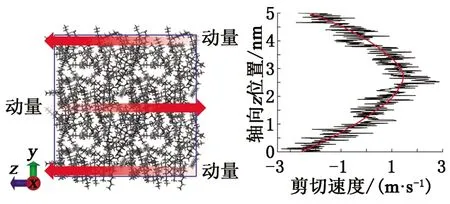
图6 Muller-Plathe反向非平衡分子动力学 方法计算黏度Fig.6 Muller-Plathe non-equilibrium molecular dynamics method for calculating viscosity
考察不同分子力场下原油组分体系的黏度特征。利用LAMMPS软件中的Muller-Plathe反向非平衡分子动力学[26]方法进行黏度计算,该方法的原理是动量在模拟盒子的两个不同层中的原子之间交换,从而在体系中产生剪切速度,根据剪切速度曲线(图6)获得体系中粒子的动量,基于线性拟合求解剪切速度曲线的斜率,进而计算体系的剪切黏度。本文中沿z轴方向对模拟体系施加动量交换,进行6 ns的分子动力学模拟,前4 ns用于体系驰豫,后2 ns用于统计计算体系的剪切速度和黏度。由于非平衡分子动力学模拟基于NVT系综开展,其无法直接控制体系的压力,为了保持稳定的流体状态,在构建模型时预先将各原油组分体系的密度设置为温度分别为373、393、413、433、453、473 K,压力为80 MPa条件下对应的密度,因此黏度验证部分只考察温度变化对原油组分黏度特征的影响,黏度验证标准仍然采用AP1700物质物性平台提供的数据。
分子动力学模拟的黏度结果见图7。相比于密度模拟,黏度模拟在非平衡态下进行,因此模拟结果存在一定的波动,但黏度总体上仍有随温度升高而下降的趋势。通过对比各个分子力场下的模拟结果可以发现,OPLS力场的计算结果与AP1700物性数据最为接近,其他3种分子力场则存在较大的偏差。根据模拟结果统计了5种原油组分在4种分子力场下的平均误差,OPLS力场为15.96%,COMPASS力场为48.53%,CVFF力场为48.61%,PCFF力场为50.15%。黏度计算结果表明,OPLS力场可以较为准确地反映模拟对象的黏度特征。
通过密度和黏度两个典型物性参数的对比可以看出,OPLS力场的模拟结果与参考数据间的平均误差最小、吻合度最高,其不仅适用于平衡态的密度模拟,也较为适用于非平衡态的黏度模拟,并且对不同类型的原油分子均表现出良好的适用性。因此OPLS力场可以被确认为是最适用于深层高温高压条件下进行原油物性模拟的分子力场。
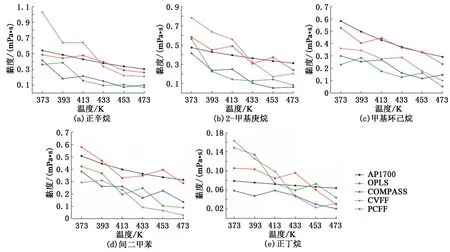
图7 样品的黏度特征Fig.7 Viscosity characteristics of samples
4 基于从头算分子动力学的OPLS力场验证
根据分子力场的势能表达式,分子力场由分子间相互作用和分子内相互作用两大部分构成。其中分子间相互作用取决于范德华相互作用(原子间距)和静电相互作用(原子电荷),这一项可以较为准确地被势函数表达;分子内相互作用的精确描述是决定力场准确性的关键,其包括原子间的键伸缩势能(键长)、键弯曲势能(键角)和二面角扭转势能(二面角)。虽然OPLS力场在计算典型原油组分的密度和黏度中表现理想,可能表明OPLS力场在模拟中所表达的分子内作用对应最优的键长、键角及二面角特征,但是该力场对分子内相互作用表达的准确性仍需从更深层次进行验证,即从更准确的电子层次进行验证。本文中采用从头算分子动力学(AIMD)模拟对OPLS力场成键特征的准确性进行验证。
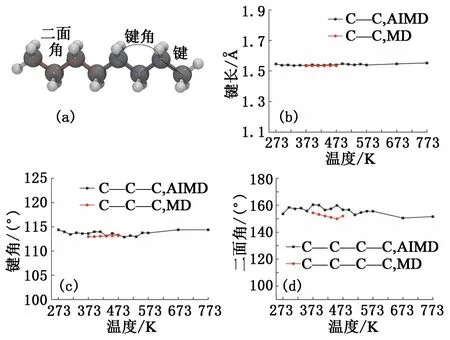
图8 正辛烷分子内碳原子间的成键及其特征Fig.8 Bonding and its characteristics between carbon atoms in n-octane molecule
从头算分子动力学模拟[27]方法将密度泛函理论和分子动力学相结合,从电子系统动力学出发计算原子间相互作用势和电子系统总能量,基于电子波函数正交化产生的“虚拟力”求解原子和电子的运动方程。AIMD方法无需预先附加力场参数,可以方便地进行分子动力学模拟,并获得准确的分子内成键信息。利用Materials Studio软件中的Dmol3模块进行从头算分子动力学模拟。由于直链烷烃、支链烷烃和环烷烃均属于饱和烃,其OPLS力场的参数一致,因此只选择正辛烷作为三者的代表进行研究,同时考察间二甲苯。模拟时的研究对象为20个分子,设置模拟时间步长为1 fs,为了更加全面地考察分子内成键特征对温度的敏感性,将模拟温度设置在273~773 K。在NVT系综下进行100 ps的AIMD模拟,统计平均最后10 ps内正辛烷和间二甲苯分子内碳原子间的成键参数,并与选用OPLS力场进行分子动力学模拟的统计结果进行对比,两种分子的成键信息如图8(a)和图9(a)所示。正辛烷分子内碳原子间的成键特征见图8(b)~(d),可以发现AIMD和MD模拟所得的键长、键角和二面角均没有随温度的改变而产生明显的变化,表明正辛烷分子的成键特征与温度条件并不敏感,其成键特征在深层环境条件下较为稳定。对比MD和AIMD的模拟结果,MD模拟下的键长和键角特征与AIMD模拟所得的结果很接近,吻合度较高,但二者的二面角特征存在些许偏差,AIMD模拟的平均二面角角度为156.15°,MD模拟的平均二面角角度为152.07°。主要原因在于AIMD模拟时的对象个数较MD模拟更少,正辛烷分子的直链结构相对保持较好,而在MD模拟体系中包含了数百个正辛烷分子,模拟过程中分子间存在更多的碰撞和缠绕,导致二面角的平均值有所降低。
间二甲苯分子内碳原子间的成键特征见图9(b)~(d)。与正辛烷分子的成键特征类似,间二甲苯在AIMD和MD模拟后所得的键长、键角和二面角特征也没有随温度的改变产生明显的变化,并且两种模拟所得的成键特征极为接近。由图9(d)看出,间二甲苯分子的二面角特征并未像正辛烷分子那样在两种模拟下产生一定的偏差,间二甲苯的碳原子间二面角均值在173°,这是由于间二甲苯分子中的苯环结构刚性较强,相邻碳原子间只存在小幅振动,不会产生较大的扭转。
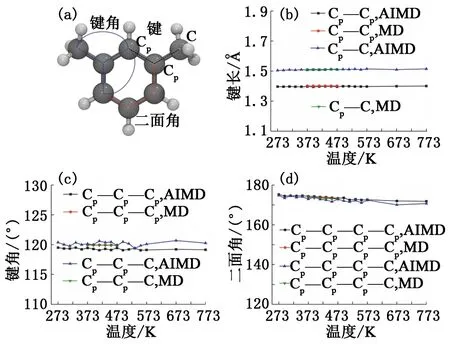
图9 间二甲苯分子内碳原子间的成键及其特征Fig.9 Bonding and its characteristics between carbon atoms in m-xylene molecule
综上所述,在当前的深层油藏条件下,AIMD与MD模拟所得的成键特征没有随温度改变而产生显著变化,说明分子的成键性质对温度条件不敏感。OPLS力场设置下,两种模拟所得的成键特征吻合良好,证实OPLS力场在分子动力学模拟中具有较高的可靠性。
5 结 论
(1)采用OPLS力场模拟得到的密度和黏度结果与AP1700参考数据最为接近,且该力场对不同类型的烷烃分子均表现出较好的适用性,OPLS力场可以被优选为最适宜在深层高温高压条件下进行模拟的分子力场。
(2)原油分子的成键特征对温度不敏感,并且OPLS力场下MD模拟的成键特征能够较好地吻合AIMD的模拟结果,证实OPLS力场的可靠性,是最适用于深层高温高压条件下进行原油模拟的分子力场。
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
高中数理化(2023年6期)2023-08-26 13:28:24
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
北京大学学报(自然科学版)(2022年4期)2022-08-18 06:58:18
中国音乐学(2022年1期)2022-05-05 06:48:46
华南师范大学学报(自然科学版)(2021年2期)2021-04-23 05:05:36
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
分析化学(2019年3期)2019-03-30 10:59:24
石油炼制与化工(2018年11期)2018-11-13 08:01:38
